时隔9年,美国心脏协会(AHA)和美国心脏病学会(ACC)终于更新了肥厚型心肌病(Hypertrophic Cardiomyopathy, HCM)诊疗指南。自从60年前第一次描述HCM这个疾病,其就有过许多历史性命名,包括特发性主动脉瓣下狭窄、肥厚型梗阻性心肌病等。由于有1/3 HCM患者左室流出道不存在梗阻,因此推荐命名为HCM(有或无流出道梗阻)。新指南同样不支持有些地区将HCM命名延伸至系统性疾病或继发性因素所致左室肥大,认为这样命名容易造成混淆,因此其它疾病或系统性疾病所致的左室肥厚不能诊断为HCM。由于HCM与其它病因所致的左室肥厚会有交叠,故需要更多的临床手段来鉴别。新指南共更新133条推荐,包括以下6大类内容:共同决策,大型HCM专业中心角色,诊断、初步评估和随访,心源性猝死(SCD)风险评估与预防,HCM管理,患者生活方式指导。新指南将患者在共同决策中列在最重要的位置,强调当做出复杂、改变生活方式的医疗决策时,必须把患者生活方式的选择和偏好纳入重点考虑。因此,它也是一部充满人文关怀的新指南。
HCM概述
HCM被认为是一种单基因心脏病,编码心肌肌小节(或肌小节相关结构)蛋白的8个基因中有1个或多个基因突变可致左心室肥厚(LVH),约30%-60%的HCM患者具有可识别的致病基因突变或可能的致病性遗传变异。
在HCM和致病性肌小节基因突变患者中,β-肌球蛋白重链基因(MYH7)和肌球蛋白结合蛋白C基因(MYBPC3)突变是最常见的两种致病基因突变(约占70%)。而其他基因(TNNI3、TNNT2、TPM1、MYL2、MYL3、ACTC1)突变各占一小部分(1%-5%)。基因突变个体的每一个后代都有50%的机会遗传变异,虽然有致病性基因突变的家庭成员发展成临床HCM的可能性很高,但每个个体的发病年龄是有差异的。
HCM是全球范围内常见的常染色体显性遗传性心脏病。虽然女性的疾病诊断率低于男性,但从遗传学角度来讲,男女患病机率是相同的。HCM的患病率取决于是否考虑亚临床病例,且取决于人口中的年龄和种族比例。据报道,美国年轻人中不明原因无症状性心肌肥厚的患病率为1:200-1:500。根据医保数据,美国成年人有症状的心肌肥厚发病率<1:3000。考虑到人群中存在大量无症状患者未被筛查发现,HCM的实际患病率要高很多。出现症状、发生心脏事件、检测到心脏杂音、常规检查心电图时发现异常或家族谱系筛查时心脏影像异常,都是怀疑HCM并采取进一步检查的契机。
HCM是特指一类由于肌小节蛋白编码基因(或肌小节蛋白相关基因)变异,或病因不明的以左室肥厚为特征的心脏疾病,需排除有明确证据证实的其它心脏、系统性或代谢性疾病导致左室肥厚的情况。且本指南从定义上将HCM拟表型(HCM phenocopies)排除在HCM临床定义之外。
成人HCM的临床诊断可通过影像学检查来确定。二维超声心动图或心脏磁共振成像显示左心室任何部位的最大舒张末期厚度≥15 mm,且没有其它导致心肌肥厚的原因即可诊断。如果基因检测阳性或者家庭成员中有HCM患者,则13-14 mm的心肌肥厚也可以诊断HCM。
对于儿童来说,诊断标准需根据体型和生长情况调整。儿童HCM的传统诊断标准是左心室壁厚度增加超过同年龄、同性别和同体表面积儿童左心室壁厚度平均值加2个标准差(或Z值>2)。
几乎任何形式的左室肥厚均可见于HCM,其最常见的部位是前间隔基底段与左室前壁。部分患者左室肥厚仅局限于1-2个左室节段,且左室质量正常。虽然收缩期二尖瓣前向运动(SAM)和左室高动力状态常见于HCM,但不是临床诊断的必要条件。许多其它形态异常不能诊断HCM,但可能是该病表型的一部分,包括乳头肌肥大和顶端移位、心肌隐窝、乳头肌直接插入二尖瓣前叶(腱索消失)、二尖瓣瓣叶伸长、心肌桥和右心室(RV)肥大。
与2011年指南相同,新指南列出的HCM主要病理生理学机制共包括5个方面:LVOTO,舒张功能不全,二尖瓣反流,心肌缺血,自主神经功能障碍。但对于特定的HCM患者,临床表现可能会是其中一种病理生理机制为主,或是多种机制共同作用的结果。因此,在遇到特定的HCM患者出现无法单纯用LVOTO来解释的临床症状或表现时,应该考虑可能是其它因素参与的结果。
尽管HCM患者的寿命可能与正常人相近,并且没有明显症状也无需特殊治疗,但另一部分HCM患者却是面临重大后果。到目前为止,发现为数不少的HCM患者直到60岁仍几乎没有伤残。然而,一个多中心注册研究表明,在患有致病性肌动蛋白基因变异和年轻时就已确诊的患者中,HCM导致死亡、HF、中风、室性心律失常、房颤(AF)的风险更大。
在以转诊为基础的HCM患者中,有30%-40%会发生不良事件,包括:1.猝死;2.由于LVOTO或舒张功能障碍导致的进行性限制性症状;3.与收缩功能不全相关的心力衰竭症状;4.有卒中风险的房颤。
诊断、初步评估与随访
对HCM的临床评估可通过以下方式触发:确定HCM的家族史,症状包括心脏事件、心脏杂音、超声心动图、12导联心电图等。应全面评估患者心脏相关的病史,包括3代以内的家族史以及全面的体格检查(包括诸如Valsalva、蹲下站立、被动抬腿或步行等)。当发现有意义的症状体征后,应进行心电图和心脏影像学检查等以鉴别左室肥厚。
心脏影像学检查在HCM的诊断和治疗中起着至关重要的作用。超声心动图是适用于大多数患者的心脏影像学检查方式,心脏磁共振成像可提供补充信息,并可应用于超声心动图无法确诊的患者。从心脏影像学检查中可获得诊断(或排除诊断)、病情评估以及心脏结构和功能评估(例如收缩、舒张或瓣膜功能不全)的重要信息。可识别动态LVOTO(包括二尖瓣的整体性能)是超声心动图的关键优势。最大室壁厚度、心腔大小、收缩功能和是否合并左室心尖部室壁瘤都可用于判断病情的严重程度以及SCD危险分层。
心血管磁共振成像(CMRI)可对心脏进行高空间分辨率和全层断层成像,并可注射造影剂进行延迟后强化(LGE)来评估心肌纤维化。CMRI的这些特点非常适合表征HCM的各种表型,提供诊断,风险预测和间隔切除的术前计划。由于这些原因,CMRI是评估HCM患者的重要辅助成像技术。
CMRI具有独特的优势。因为可以在血液和心肌之间产生具有鲜明对比的图像,从而可以提供高度准确的左心室(LV)壁厚度的测量值、LV和RV腔室大小、LV质量、收缩功能的可靠定量,并且可以识别出超声心动图不能很好观察的LVH。CMRI也使我们对于各种形态异常有更多的了解,包括LV心尖动脉瘤以及导致LVOTO的二尖瓣和瓣下结构异常装置,这些发现可能会影响治疗策略。此外,广泛的LGE意味心肌纤维化程度,这代表一种无创性标志物,增加了潜在的危及生命的室性心动过速和心力衰竭(HF)伴收缩功能障碍的风险。目前认为,设备缺乏、费用、起搏器或ICD禁忌、严重肾功能不全、患者因素(小儿年龄、全身麻醉、镇静、幽闭综合征或体型)是特定一部分人群无法完成CMRI的原因。
心脏CT提供出色的空间分辨率,可以明确LV结构(包括肥大方式、室壁厚度测量、主动脉瓣下隔膜和心内血栓)和功能。小型研究表明CT具有评估心肌纤维化的能力,尽管有可能增加放射线暴露。除了心肌结构,CT还可以评估冠状动脉的解剖结构,包括冠状动脉的狭窄和起源异常。CT的缺点是使用放射线和放射性碘造影剂,而且与超声心动图相比其时间分辨率较差。
同时使用12导联心电图和动态监护对于HCM患者是必要的。12导联心电图可以提示有关LVH和复极异常以及心律不齐(包括心动过缓和心动过速)的信息。它还提供了有关传导异常的信息,这些信息可能在初始评估或随访中出现。在评估SCD风险时动态监测24-48小时是必要的。持续的监视对于确定发生症状的原因或诊断AF最有用。
在过去的60年中,梗阻性HCM患者的血流动力学特征和评估已得到公认。超声心动图仍是无创评估HCM患者动态流出道梗阻的金标准。因此,没有强有力的证据在梗阻性HCM患者常规评估中进行侵入性血流动力学评估或在HCM普通人群中常规行冠状动脉造影。仅当患者决策性的诊断信息无法从临床和非侵入性影像学检查中获得,才借助于侵入性血流动力学评估。因此,所选定的患者亚群才能从这些侵入性检查中受益。在这种情况下,操作者必须经验丰富,并使用适当的导管,同时避免诸如导管卡顿之类的危险,这一点至关重要。
有证据表明,运动负荷检查,特别是结合使用同步分析呼吸气体(即心肺运动测试(CPET)),对于HCM患者是安全的,并可提供有关功能受限的严重程度和机制的信息。由于静息心电图和室壁运动异常,运动试验在评估心肌缺血方面的价值有限。使用单光子或正电子发射断层扫描的心肌灌注成像显示,>50%的患者存在心肌灌注异常,其中大多数患者没有明显的心外膜冠状动脉病变(CAD)。
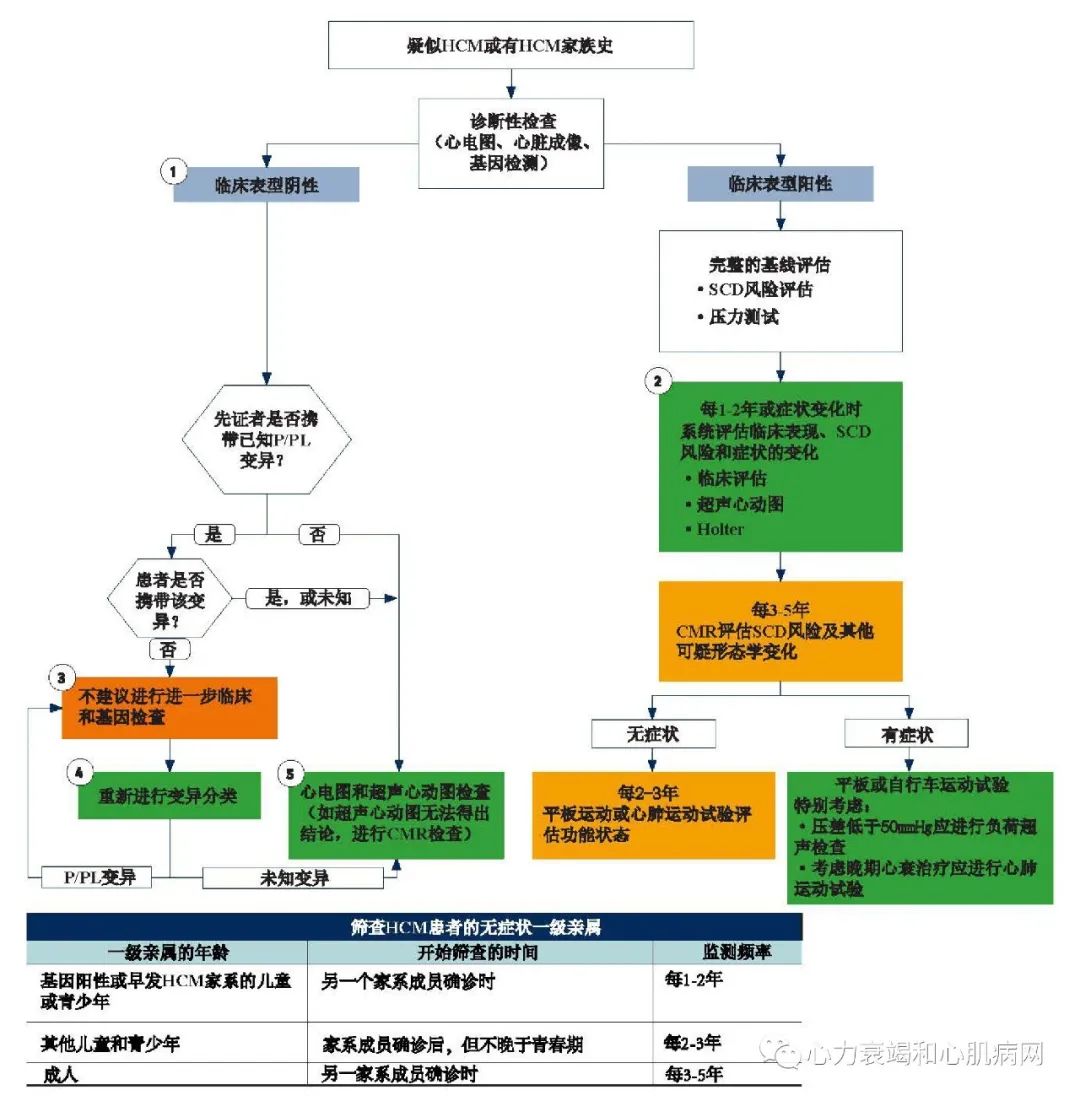
图1 HCM的评估与筛查
基因检测在HCM患者及其亲属的诊断和管理中起着重要作用。大多数情况下,HCM为常染色体显性遗传,其后代有相同致病基因突变的概率为50%。与HCM患者讨论基因检测的作用是临床决策的标准部分,包括由专业人员在检测前后进行遗传咨询,有必要收集多代人(最好大于3代)HCM家族史及可疑的SCD事件;同时应考虑到心理、社会、法律、道德和职业等因素在遗传性疾病获得中的重要影响。理想情况下,遗传评估应在多学科的HCM中心进行,且该中心在遗传咨询和检测方面应具备一定经验。
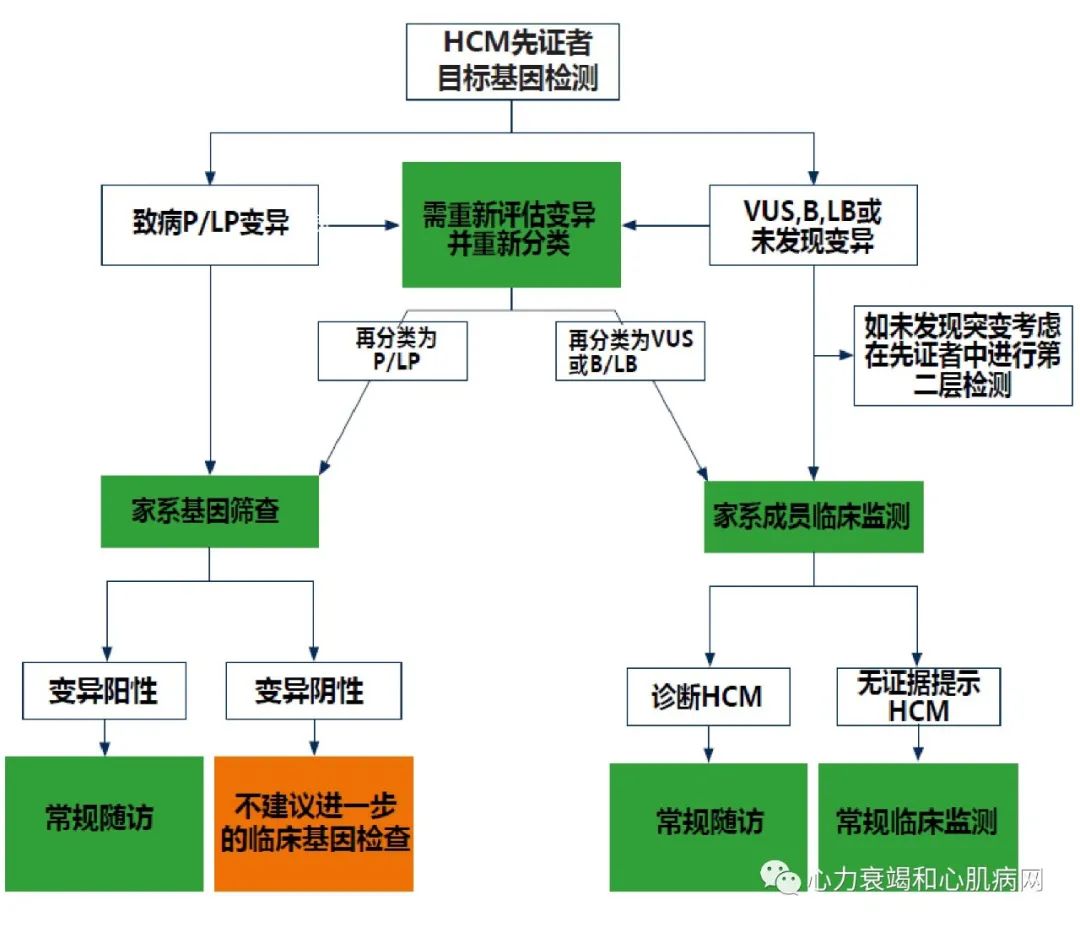
P/LP:致病/可疑致病突变;B/LB:良性/可疑良性突变;VUS:未知意义的突变
图2 HCM基因检测及家系筛查指南推荐
“基因型阳性,表型阴性”是指携带致病或可疑致病的突变基因,但无症状及影像学左室肥厚证据的个体。这些人也被描述为临床前HCM。他们需要持续监测以发现HCM发展至临床阶段,尽管从遗传诊断到发展至临床HCM的时间在家庭成员之间和不同家庭之间有较大差异。研究报道HCM患者可出现多种结构和功能异常,如:心肌应力改变,左室舒张功能异常,心肌隐窝,二尖瓣前叶异常,异常肌小梁,心肌瘢痕形成,心电图异常,血清NT-Pro BNP异常但无左室肥厚。然而,亚临床阶段结构和功能异常的意义尚不清楚。因此,通常治疗方式的选择不单基于上述这些特征。
SCD的风险评估及预防
HCM是造成北美洲年轻人SCD最常见的原因,也是这一遗传性心脏病最常见及最严重的并发症。年轻HCM患者SCD发生风险高于老年患者。儿童期HCM患者5年SCD事件累计发生率约8%-10%。目前尚未发现性别、种族间SCD事件发生风险存在差异。
新指南认为5年猝死风险率公式没有纳入新的猝死风险标志物如LVEF<50%、心尖室壁瘤、钆延迟强化。如果存在上述情况之一,则5年猝死风险被低估。新指南I类推荐建议一旦诊断后立即进行综合、系统的无创风险评估,此后每1-2年评估1次。评估指标包括:心脏骤停或持续室速的个人史,怀疑与心律失常相关晕厥,近亲属HCM相关的过早猝死、心脏骤停和持续室速,最大室壁厚度、EF、LV心尖室壁瘤,连续心电监测有非持续性室速。
过去数十年的研究聚焦SCD风险因素的探索,并依此对患者进行危险分层进而发现需要进行ICD植入预防SCD的高风险患者。随着风险分层策略的实施与ICD在临床中日益广泛的应用,疾病相关的死亡率已有显著的下降。风险预测评分推动了5年SCD事件的个体化风险预测,这一策略也完善了患者的风险分层及成人ICD植入的诊疗决策。随着SCD风险预测体系的不断发展和新的风险预测标志物的出现,运动血压异常反应已不再应用于预测HCM患者SCD风险。
目前临床常用的评估患者SCD风险和评估初级预防ICD植入效果的非侵入性指标,是个人史与家族史的情况及非侵入性检查(包括超声心动图、动态心电图监测及心脏磁共振成像)选择而来的。由于SCD的风险长期存在,阶段性评估是HCM患者长期管理不可或缺的一环。
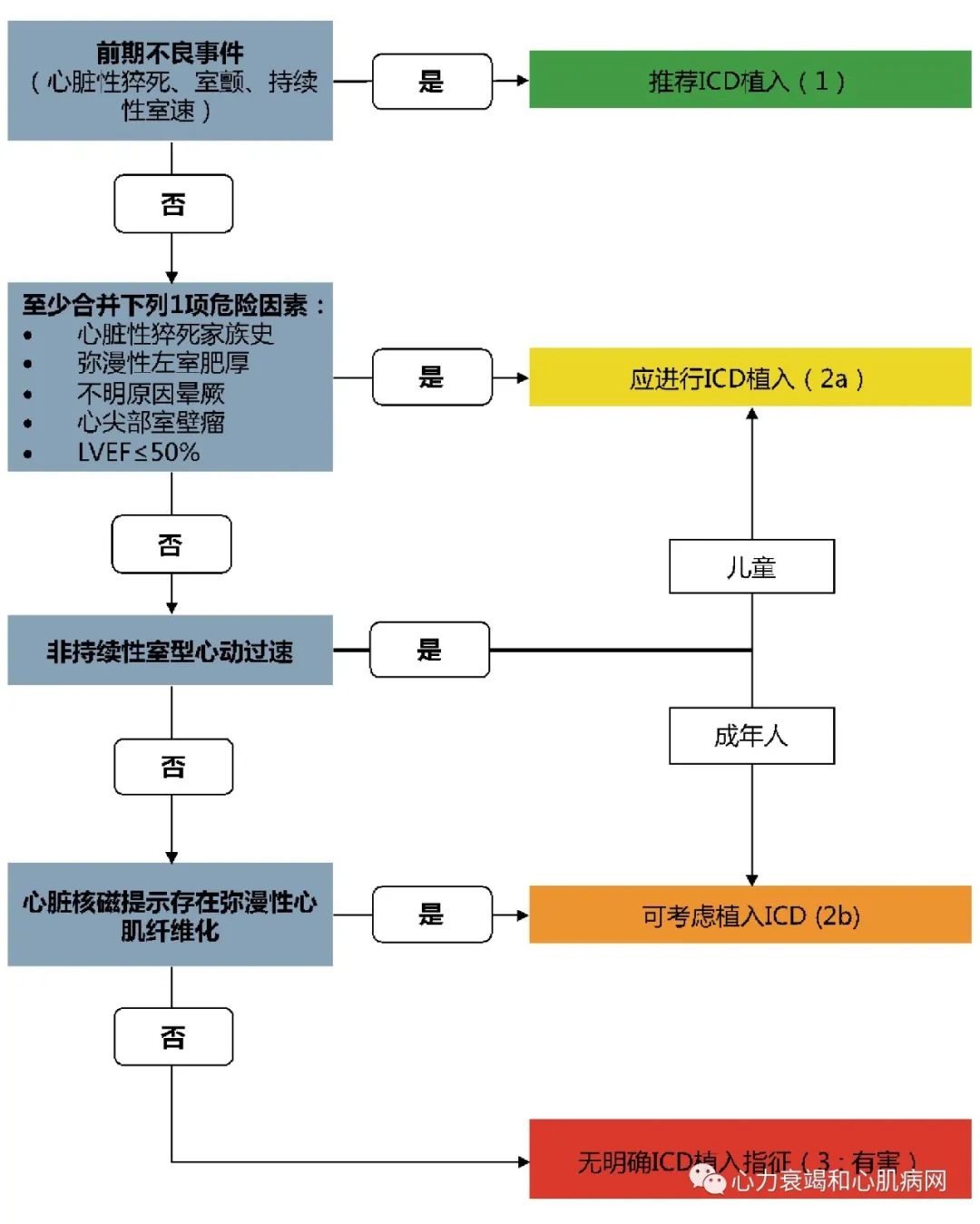
图3 HCM患者ICD植入流程图
阜外医院心力衰竭中心、HFCU简介
阜外医院心力衰竭中心、心力衰竭重症监护病房成立于2002年,是中国首个以心力衰竭、心肌病为诊治特色的临床医疗中心。作为国家级诊疗中心,在张健教授、张宇辉教授的带领下,以及所有医护人员的积极推动下,重点工作在于多学科协作诊治疑难和危重心力衰竭,主要收治各种心血管疑难、危重症,及各种原发性和继发性心肌病,包括扩张型心肌病、肥厚型心肌病、限制型心肌病、肿瘤心脏病等;注重疑难心脏病的病因诊断和精细化治疗, 率先在国内开展肿瘤心脏病及心脏淀粉样变的诊疗及科研工作。在2020年12月7日维万心通过进口药检测报告国内开售当日,即为一名确诊ATTR-CM患者及时给与维万心的治疗。此外,病房兼收治TAVI术前术后管理、重症心脏病的冠脉介入治疗以及冠脉、肺动脉、起搏器、射频消融介入术后并发症等疾病。
心力衰竭中心是一流的心力衰竭科研国家级团队,先后主持“十二·五”、“十三·五”国家科技支撑项目、国家自然科学基金等多项有关心力衰竭的科研项目,作为项目负责人参加了10余项国际大规模多中心RCT研究,创办了第一本心力衰竭和心肌病领域的专业学术期刊《中华心力衰竭和心肌病杂志(中英文)》,组织撰写并发表《中国肥厚型心肌病管理指南2017》、《心力衰竭容量管理中国专家建议》、《中国心力衰竭患者离子管理专家共识》等10余项专业指南和诊疗规范。
作为中国心力衰竭和心肌病领域的领军团队,阜外医院心力衰竭中心、HFCU将继续致力于建立、推广及普及心力衰竭标准化诊疗及心肌病精准诊疗流程,以改善广大心力衰竭患者的长期预后及生活质量。
参考文献:Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, Evanovich LL, Hung J, Joglar JA, Kantor P, Kimmelstiel C, Kittleson M, Link MS, Maron MS, Martinez MW, Miyake CY, Schaff HV, Semsarian C, Paul S. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Nov 20:CIR0000000000000938. doi: 10.1161/CIR. 0000000000000938. Epub ahead of print. PMID: 33215938.
供稿:庄晓峰 翟玫
审校:张宇辉
责编:赵雪梅 梁琳 赵朗 黄博平



