
心衰患者室性心律失常的发生率 80%~95%,非持续性室速占30%~40%。除原有心律失常外,相当一部分在心衰进展到一定程度时出现。Framingham研究50%~60%的心衰患者死于猝死和心律失常。心衰患者SCD中50%~75%与室性快速性心律失常有关。
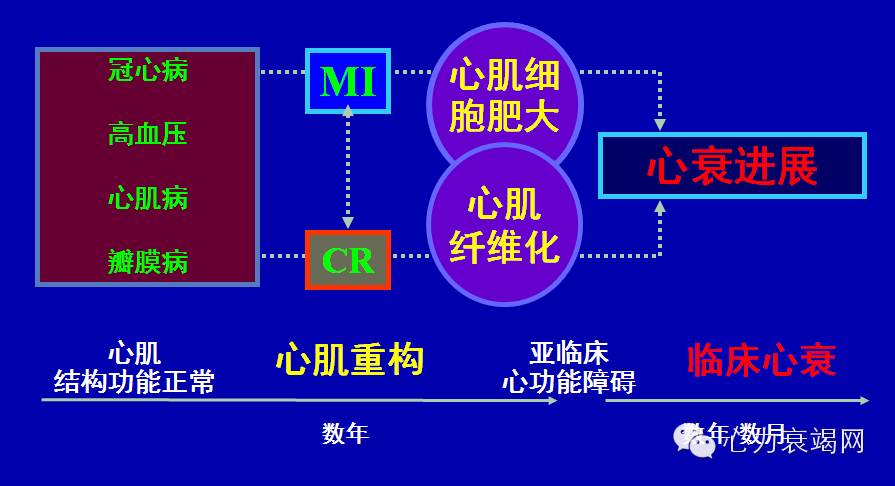
心 肌 重 构
在内源性或外源性因素影响下,心肌结构和功能不断调整,左心室和(或)右心室空间构象和生物效应改变,局部或大部甚至全部心肌肥厚或变薄。高血压、心肌缺血、瓣膜病和心肌病等均可引起心肌重构。
细胞水平:单个心肌细胞肥大,体积增加。间质成分如成纤维细胞增殖,胶原合成和分泌增加,伴随着心肌细胞凋亡,引起心肌组织成分发生改变.心肌细胞排列紊乱
分子水平: 蛋白合成增加. 核内基因表型模式改变,胚胎型基因β-MHC被激活,成熟型基因α-MHC表达下降;同时,促进心肌纤维化的基因表达增加,如结缔组织生长因子CTGF、转化生长因TGFβ,Ⅰ型和Ⅲ型胶原等
分子机制:多种经典信号转导通路及信号通路效应分子相互作用,形成信号网络,共同调控心肌细胞肥大、心肌纤维化以及细胞凋亡相关靶基因和蛋白的表达。主要涉及:
丝裂原活化蛋白激酶(MAPKs)
钙调神经磷酸酶(Calcineurin)/ NFAT
Janus激酶/信号转导与转录激活子(JAK/STAT)
蛋白激酶C(PKC)
磷脂酰肌醇-3-激酶/蛋白激酶B(PI3K/Akt)
Wnt/β-连环蛋白(β-catenin)和转化生长因子β(TGF-β)/Smad等
心肌重构贯穿心衰全过程
肥大的心肌细胞离子通道改变
钠通道、钙通道、钾通道包括瞬时外向钾通道(Ito) 、内向整流钾通道(Ik1)、快速延迟整流钾通道(Ikr)、缓慢延迟整流钾通道(Iks)等通道密度减少 。
心力衰竭时, INa+、Ica-L、 Ito 、Ik1、Ikr 、Iks 、I C a - L减少。
钙离子
胞内外、钙库内外有明显的浓度差(12000倍)
钙离子的特性使其易于与蛋白质形成紧密的结合
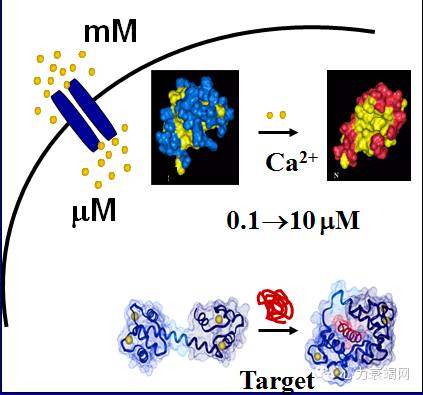

钙离子释放产生钙火花
镁离子
Mg2+是钙离子通道的阻断剂
Mg2+阻断再灌注引起的自由基形成
Mg2+降低缺血再灌注后的心肌损伤
Mg2+作为细胞辅助因子帮助心肌重建氧化代谢和补充高能磷酸化合物的储存
Mg2+-ATP复合物是肌肉收缩和舒张过程中酶促反应的底物
心衰时血镁浓度下降
镁离子的抗心律失常作用
对保持跨膜的离子平衡起重要作用
缺血期间阻止有害的钾离子外流和钙离子内流
维持细胞离子梯度的重要离子
可用于治疗心室性心动过速、快速型心律紊乱、室上性心律失常,包括心房纤颤
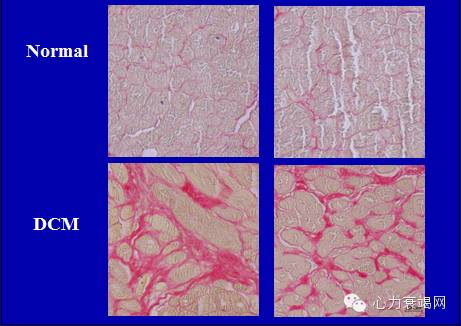
终末心衰病人心肌严重纤维化
神经内分泌系统激活
肾素-血管紧张素-醛固酮系统 RAAS
肾上腺素
B型尿钠肽 BNP
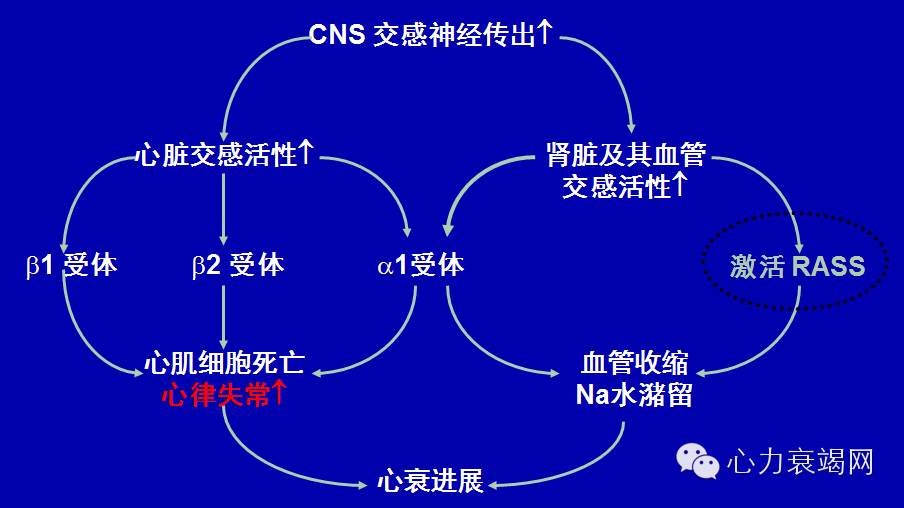
交感兴奋作用
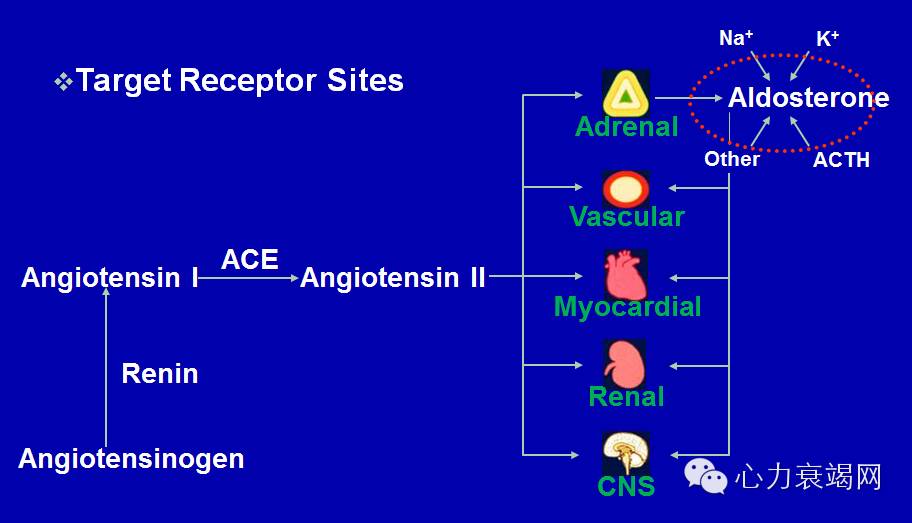
RAAS的作用
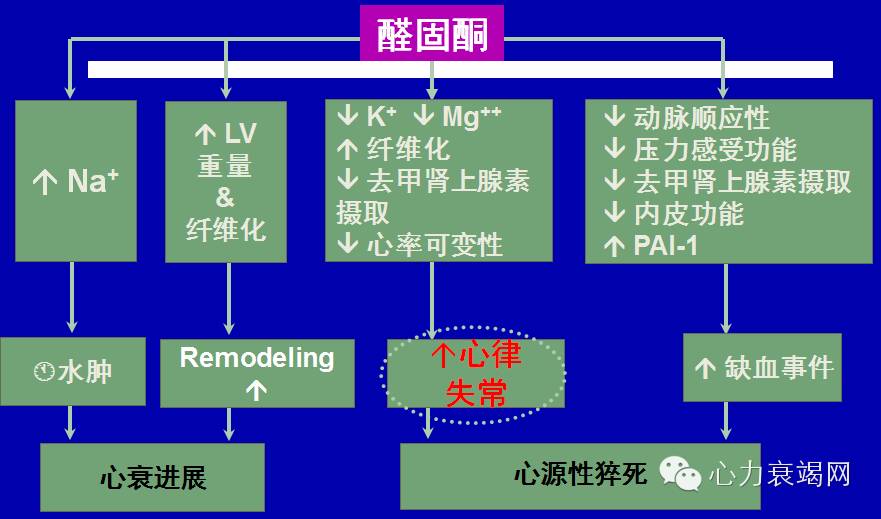
醛固酮的作用
慢性心衰合并室性心律失常的治疗基石为β受体阻滞剂和ACEI。合并室内传导阻滞则β受体阻滞剂慎重使用。合理应用利尿剂、血管活性药物改善心衰
消除诱因
心肌缺血引起者重建血运
血流动力学改变明显的室性心律失常,常为持续性室速、室扑、室颤,应立即电复律
血流动力学相对稳定:药物治疗、器械治疗、导管消融
根据胺碘酮治疗慢性心衰并室性心律失常的Meta分析结果,胺碘酮治疗慢性心衰并室性心律失常在心衰症状、室性心律失常 、心率、LVEF、 心脏猝死率方面疗效良好,具有统计学意义。
抗重构治疗
抗重构治疗以机制干预为主,特异性疗法欠缺。
RAAS 阻滞剂
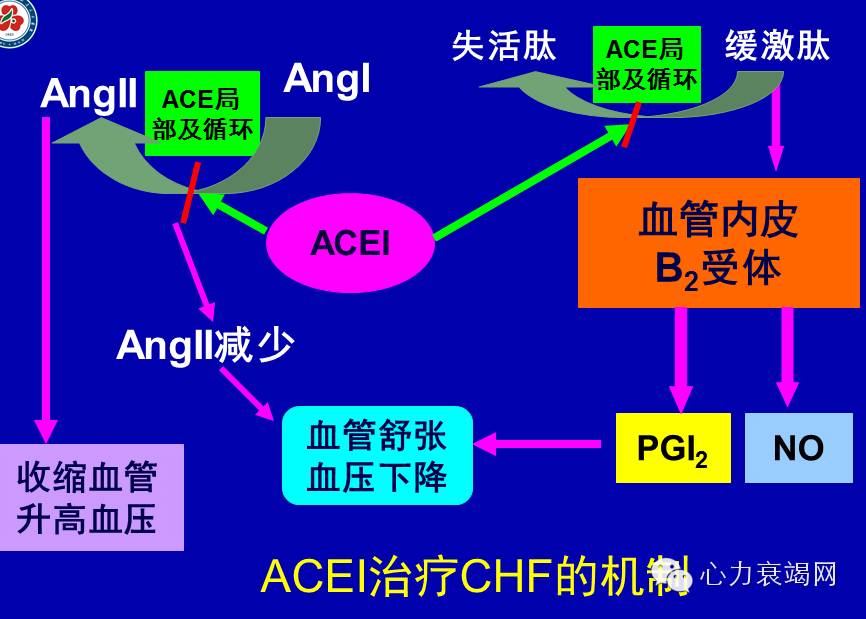
ACEI和ARB:通过抑制血管紧张素转化酶的活性以及拮抗血管紧张素Ⅱ受体结合来阻断RAAS系统.抑制血管紧张素Ⅱ介导的缩血管、促增殖、促凋亡以及促纤维化等效应 ,进而起到扩血管、逆转心室重构和改善心功能的作用。
醛固酮受体拮抗剂:心衰时醛固酮水平升高 。醛固酮过多会导致心肌胶原增生、心室肥厚和交感神经活性增强。RALES 等证明,醛固酮受体拮抗剂可显著降低总死亡率。RALES试验的一个亚组分析显示,螺内酯可抑制心肌纤维化。
β受体阻滞剂:可抑制交感神经激活对心力衰竭代偿的不利作用
心脏再同步化治疗CRT:近10年交叉研究(MUSTIC和PATH-CHF) 或平行研究(COMPANION和CARE-HF)等随机试验对CRT长期临床效果作了评估。结果显示,最佳药物治疗基础上,CRT可改善症状和生活质量,降低HF死亡率,降低全因死亡率和心源性猝死风险,逆转心室重构。有利影响长期存在(CARE-HF研究达到36个月).
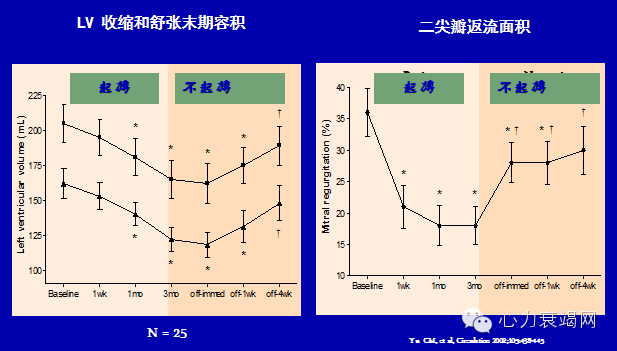
CRT逆转左室重构



