
病例分析
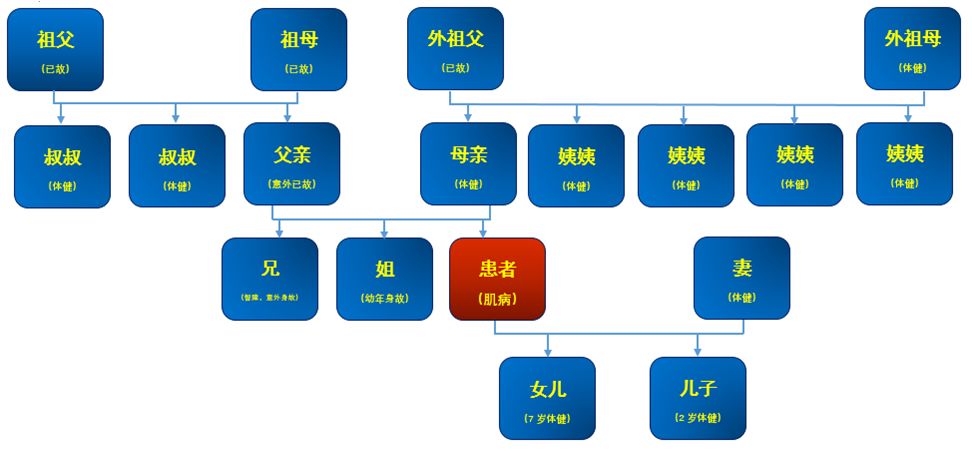
★ 患者:贾XX,男性,30岁。
★ 主诉:间断胸闷、气短、心悸2月。
★ 现病史:患者近2月于劳累及快步走时出现胸闷、气短、心悸不适,休息后上述症状缓解,每次持续约10余分钟,无夜间阵发性呼吸困难。在当地医院做心脏彩超示:心脏扩大,左室收缩功能减低。诊断为“扩张型心肌病”,给予“贝那普利、美托洛尔、呋塞米、螺内酯、地高辛”等药物治疗后症状有所缓解,入院拟进一步明确诊断及治疗。
★ 既往史:9年前诊断“肌肉萎缩”。
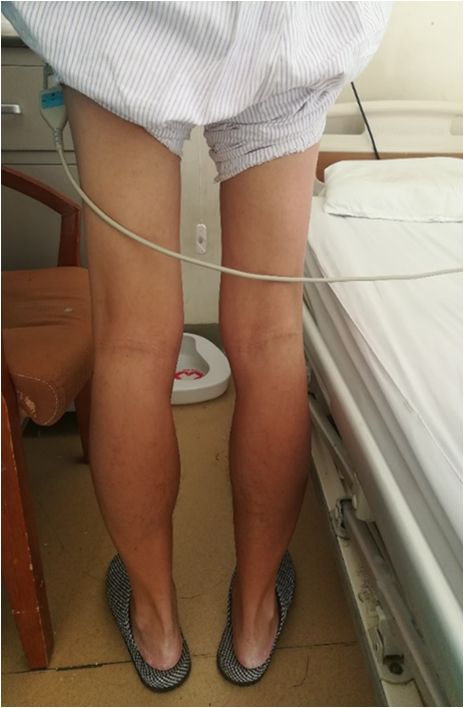
★ 体格检查:BP 100/60mmHg,双肺呼吸音清,未闻及干湿性啰音,心界左下扩大,心率80次/分,律齐,二尖瓣听诊区可闻及收缩期吹风样杂音,3/6级。双下肢近端肌肉萎缩,双侧腓肠肌假性肥大,近端肌肉肌力减退,肌张力、感觉正常,病理征(―)。
★ 病史回顾:
患者9年前(2008年12月)因“双下肢无力10年”就诊于“河北医科大学第三医院神经肌肉病区”。
临床表现:跑步慢,易摔倒,双下肢无力,易疲劳,蹲下站起、上下楼梯费力。
查体:双上肢肌力5级,下肢近端4级,远端5级。双侧腓肠肌假性肥大,余肢体肌容好,肌张力、感觉正常,四肢腱反射正常存在,病理征(―)。
实验室检查:血CK:2089 U/L。
心脏彩超示:左室稍增大,三尖瓣少量返流。
★ 病史回顾:
肌电图:呈肌源性损害。
右股四头肌活检病理:可见少量肌纤维变性、再生、坏死,肌纤维直径大小不一(15-130um),可见中心核,肌纤维分裂,结缔组织重度增生,许多肌纤维细胞色素C 氧化酶活性减低。单克隆抗体免疫组化染色:抗肌萎缩蛋白-N、-C、-R,抗肌聚糖蛋白-α、-β、-γ、-δ表达正常,抗-dysferlin蛋白表达减弱。
病理诊断:肌病(重度)。
出院诊断:股四头肌肌病。
出院医嘱:注意心脏情况,定期复查。
近9年,未行进一步诊治及随访,自觉双下肢无力症状无进行性加重。
★ 实验室检查:
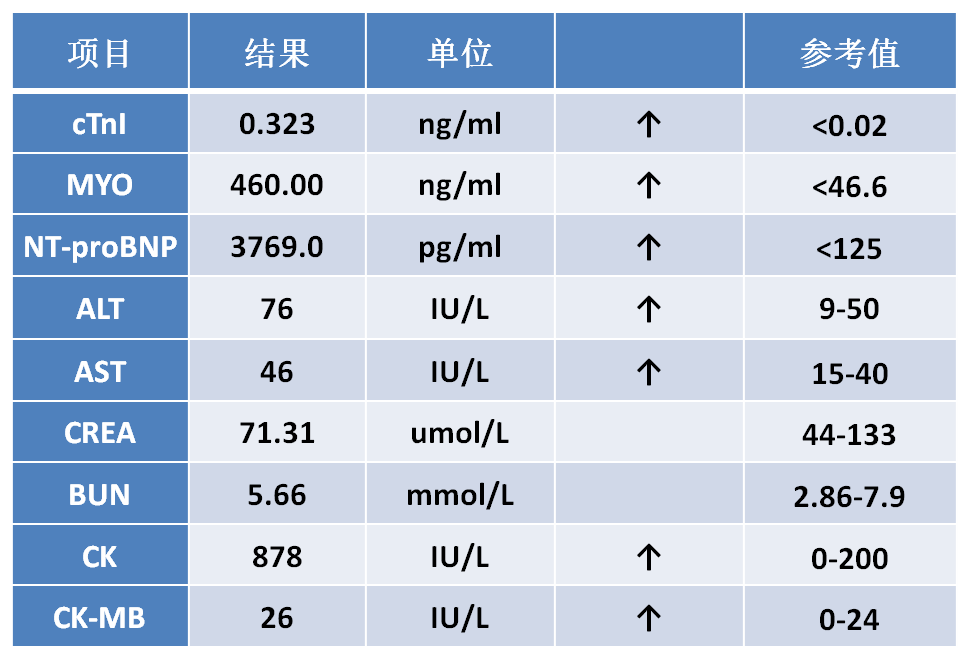
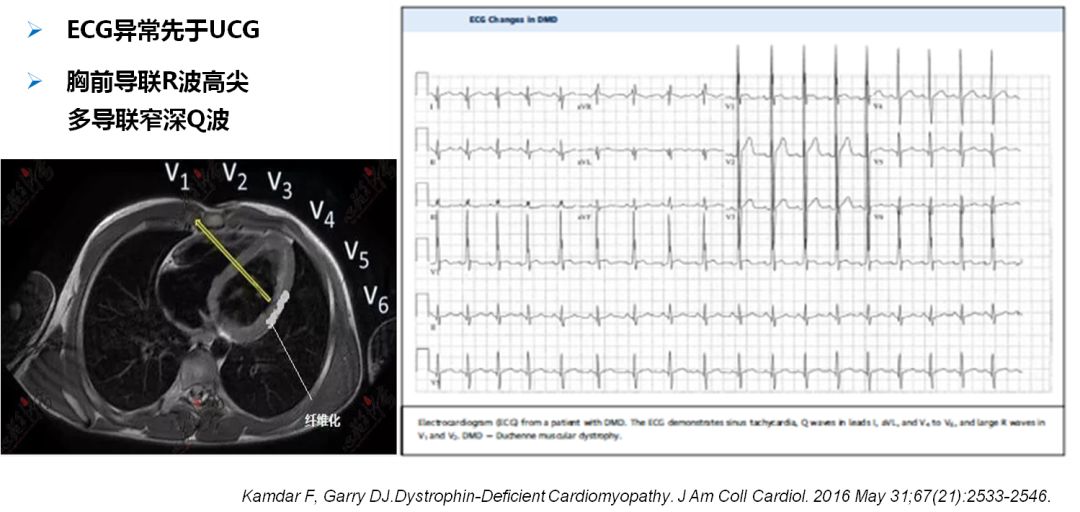
★ ECG特点:
I、avL导联Q波
胸前导联R波高尖
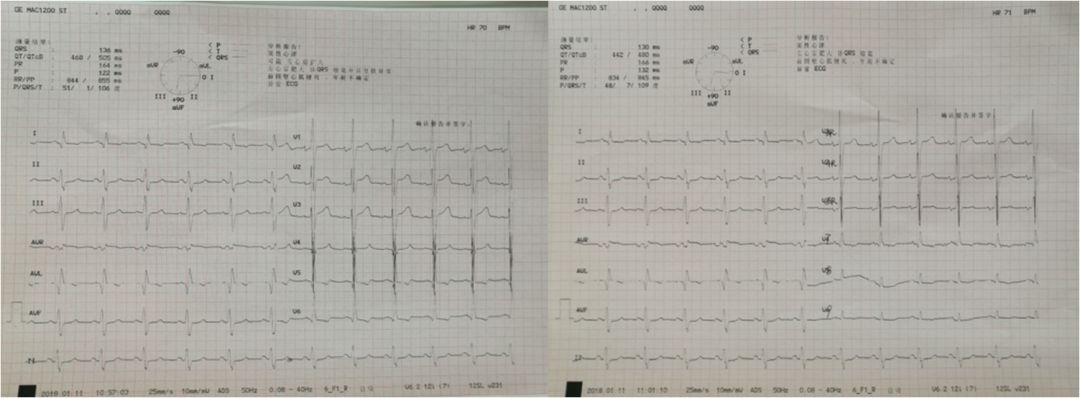
★ UCG表现:
心脏受累疾患
全心扩大(LA 55mm,LV 75mm)
二尖瓣返流(中量)
三尖瓣返流(少量)
左心功能减低(EF 37%)
肺动脉高压(中度)
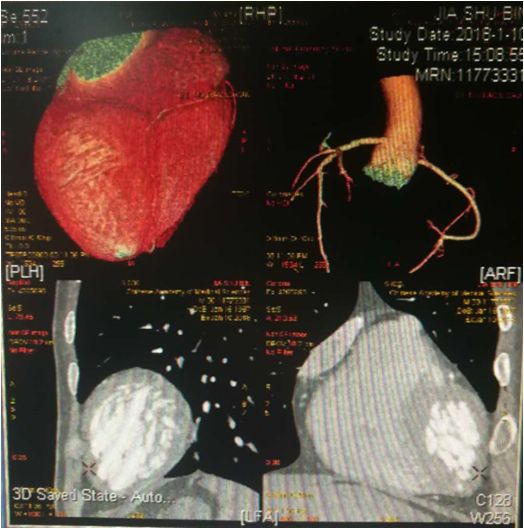
★ 冠状动脉成像:
各支冠状动脉均未见钙化;冠状动脉呈右优势型;左右冠状动脉各节段未见狭窄性病变。
左心房、室明显扩大,左心室壁普遍变薄,符合扩张型心肌病改变。
★ 诊断:
进行性肌营养不良
心脏扩大
二尖瓣关闭不全(中度)
三尖瓣关闭不全(轻度)
肺动脉高压(中度)
心力衰竭
心功能Ⅲ级
进行性肌营养不良
PMD
进行性肌营养不良定义
进行性肌营养不良(Progressive Muscular Dystrophy, PMD)是一组进行性发展的以骨骼肌变性为主要特征的遗传性疾病,许多类型已经确定为某种肌纤维膜或核膜蛋白的异常所致。
骨骼肌变性—进行性肌肉萎缩、无力;无感觉障碍
遗传性疾病—有家族史;可以进行基因诊断,基因治疗
蛋白异常—可以用免疫组化或蛋白印迹方法诊断
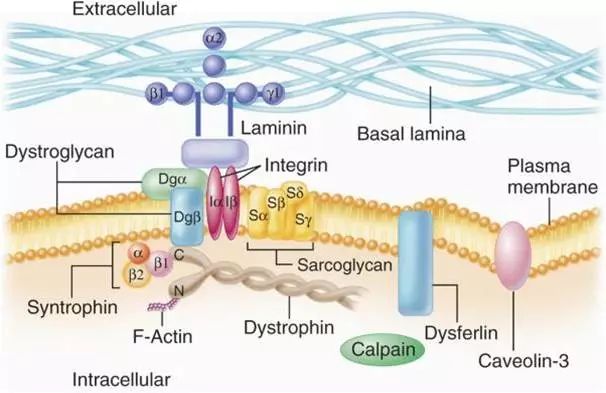
肌(纤维)膜组成
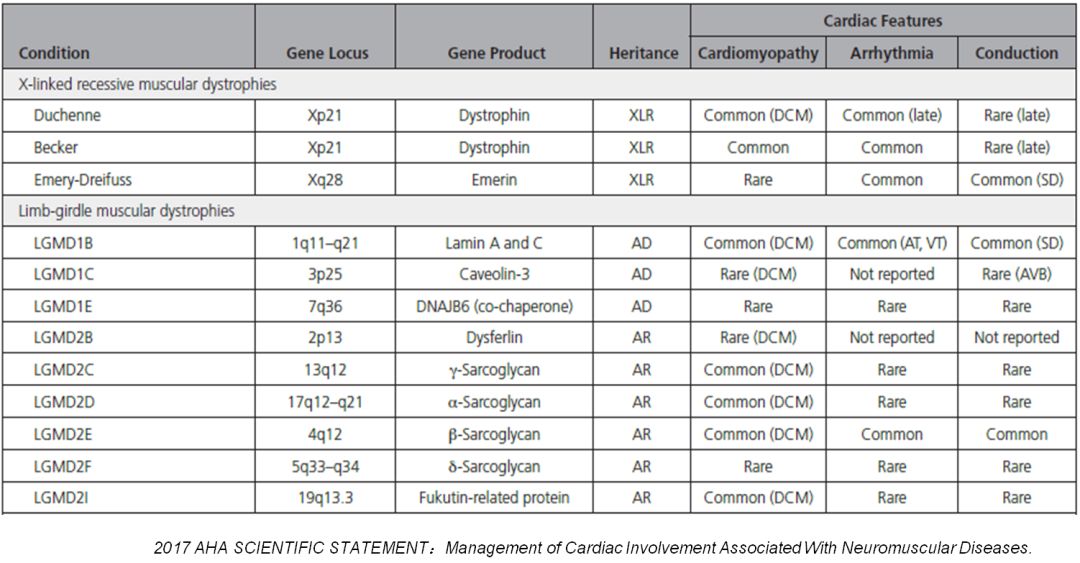
进行性肌营养不良分类

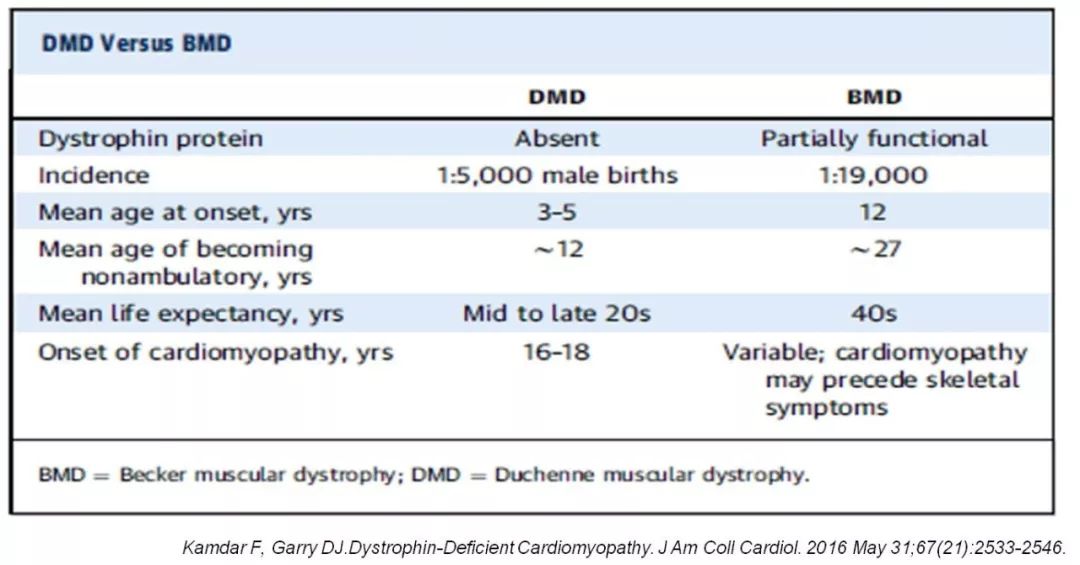
假肥大型肌营养不良
DMD/BMD
假肥大型肌营养不良
包括DMD 和BMD(Duchenne /Becker)
X 连锁隐性遗传性疾病,1/3病例为散发(基因突变)
致病基因为dystrophin (抗肌萎缩蛋白),位于染色体 Xp21
DMD:dystrophin 蛋白完全缺乏
BMD:dystrophin 蛋白量的减少
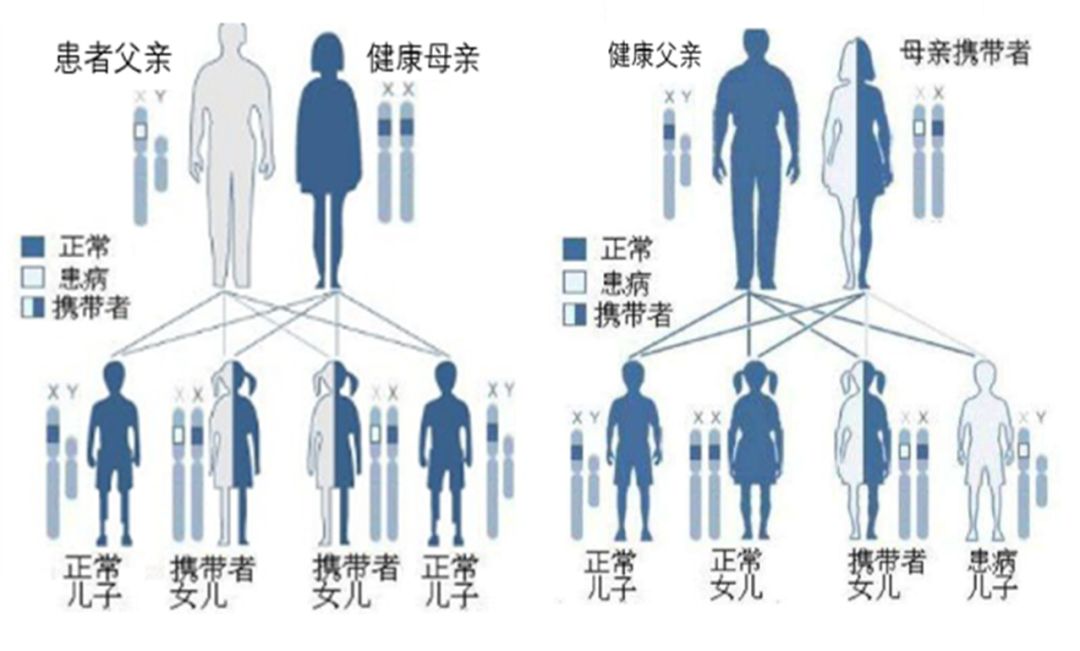
假肥大型肌营养不良遗传特点
假肥大型肌营养不良致病基因
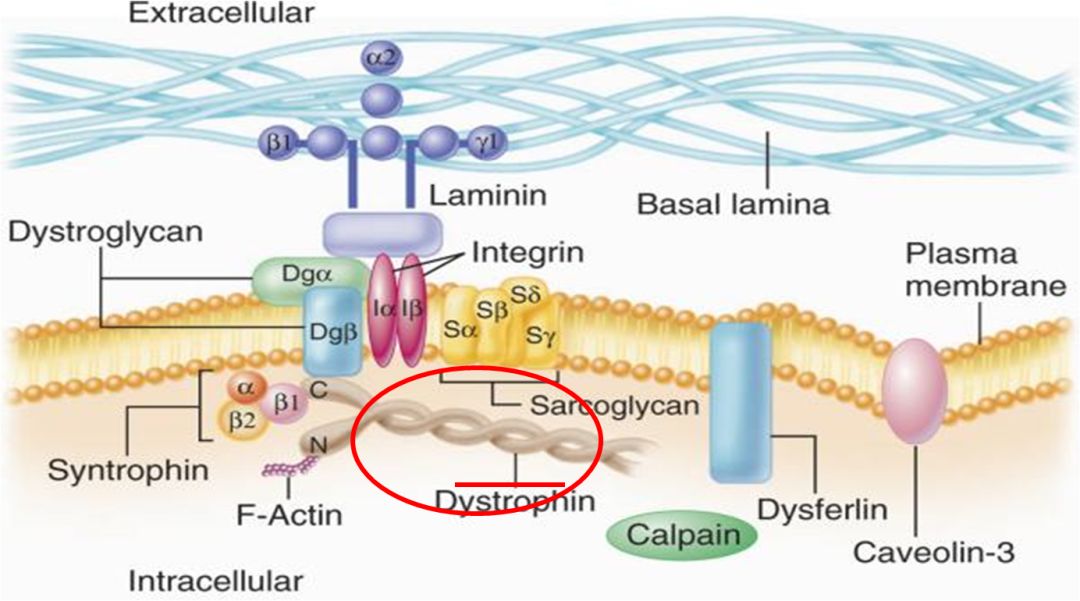
假肥大型肌营养不良致病基因
——Dystrophin(抗肌萎缩蛋白)
Dystrophin蛋白位于骨骼肌和心肌细胞膜内面,是肌纤维膜细胞骨架的成分,具有抗机械牵拉作用,能防止肌细胞在收缩过程中的损伤。
Dystrophin 与肌细胞膜和细胞外基底层之间的多种蛋白紧密结合,维系细胞膜内外物质交换和联系,保护细胞膜结构完整和稳定。
Dystrophin基因缺陷导致肌细胞膜上dystrophin 蛋白缺乏或减少使肌 细胞膜不稳定而引起肌细胞坏死和功能丧失,脂肪组织和纤维结缔组织增生。
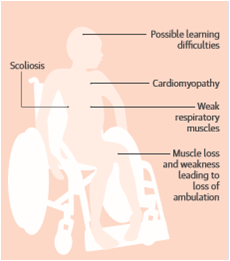
假肥大型肌营养不良临床表现
运动发育晚,跑步困难,跳跃不能,上楼梯和爬坡费力。
进行性对称性肌肉萎缩无力,近端肌肉累及早于远端肌肉,最终出现呼吸肌麻痹。
步态异常―鸭步:摇摆不稳,步基宽,易摔倒,由于脊柱过度前突,跟腱挛缩。
肌肉假性肥大:腓肠肌、舌肌等,由于肌纤维变性坏死,被纤维结缔组织取代。
翼状肩胛―前锯肌和斜方肌无力, 不能固定肩胛内缘,肩胛游离,双臂前推时尤明显。
Gower征:DMD的特有,腹肌和髂腰肌无力,仰卧起须先俯卧位,用双手臂攀附身体直立。
智力障碍:见于DMD。
其他受累系统:关节挛缩,脊柱侧突,扩张性心肌病。
假肥大型肌营养不良临床表现

假肥大型肌营养不良分类特点
假肥大型肌营养不良诊断
有家族史: XLR 遗传
典型的临床表现
血清肌酶显著升高:CK, LDH, AST, ALT
肌肉MRI:受累肌肉出现不同程度的水肿、脂肪浸润和间质增生
肌电图:肌源性损害(轻收缩时运动单位电位时限缩短,波幅降低,最大用力收缩时电位密集的病理干扰相)
肌肉活检:典型的肌营养不良特征(肌纤维大小不等,肌纤维变性、坏死和吞噬现象;肌纤维肥大、增生和分裂,可有核内移纤维;肌纤维间隙增宽,大量脂肪组织和纤维结缔组织增生);Dystrophin 抗体免疫组织化学染色:肌纤维膜不着色(DMD)/部分着色(BMD)
基因检查:PCR or point mutation screening,外显子缺失、重复、微小突变或点突变
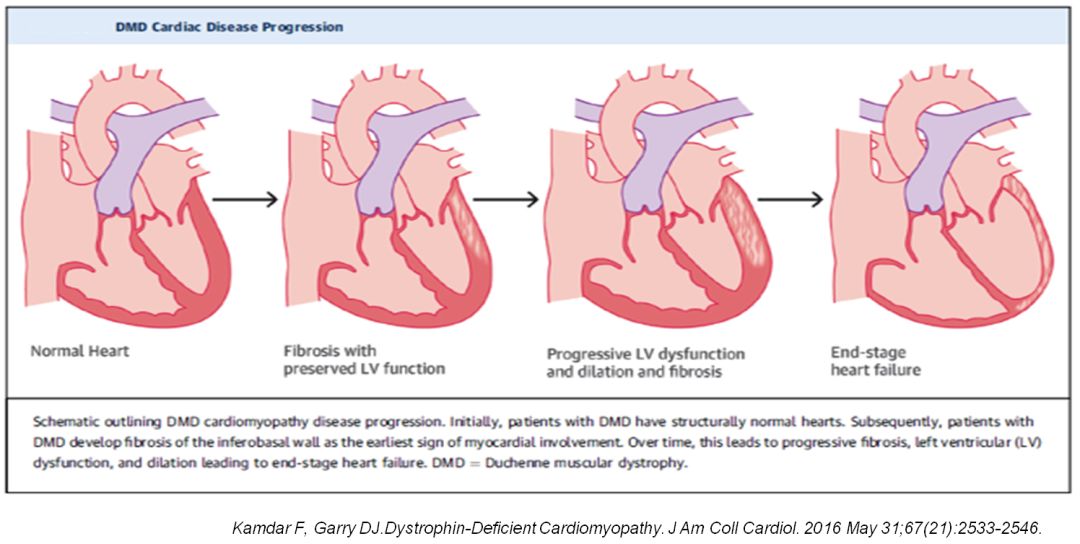
假肥大型肌营养不良心肌损害机制
Dystrophin是肌纤维膜细胞骨架的成分,使肌细胞膜和细胞外基底层之间相连,构成肌纤维收缩和松弛时肌膜保持机械性稳定的结构基础。
Dystrophin 在骨骼肌和心肌均有表达。
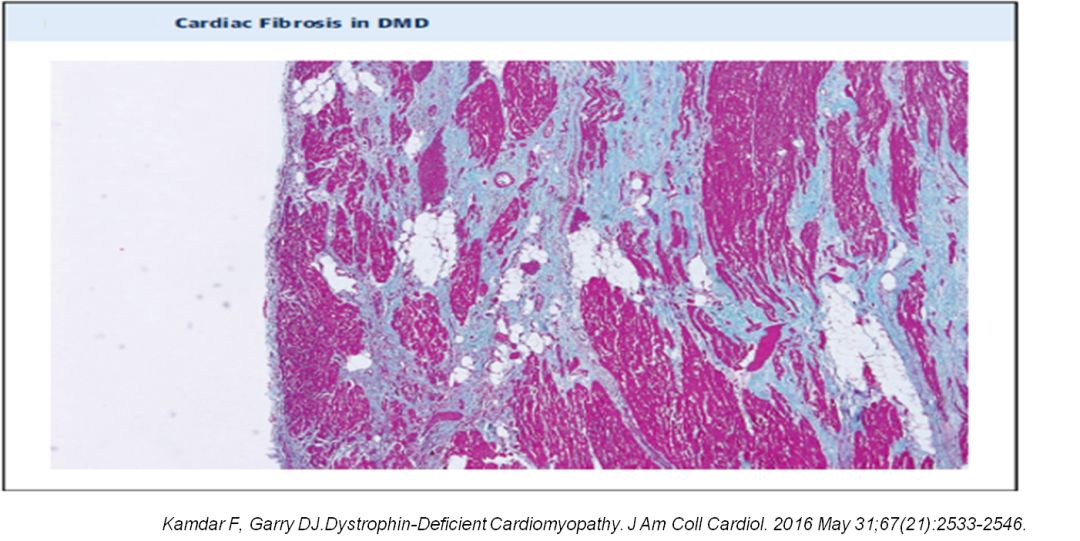
同骨骼肌Dystrophin 的缺失一样,心肌Dystrophin 的缺失,使心肌纤维化和脂肪浸润,尤其以左心室壁明显。
假肥大型肌营养不良心肌损害机制
假肥大型肌营养不良ECG表现
假肥大型肌营养不良UCG表现
二尖瓣和(或)三尖瓣关闭不全
肺动脉瓣关闭不全
左心房和(或)左心室增大
左心室射血分数明显降低
建议未发生心脏损害时,10岁以前每2年行超声心动图检查,10岁以后每年行超声心动图检查。
假肥大型肌营养不良CMR表现
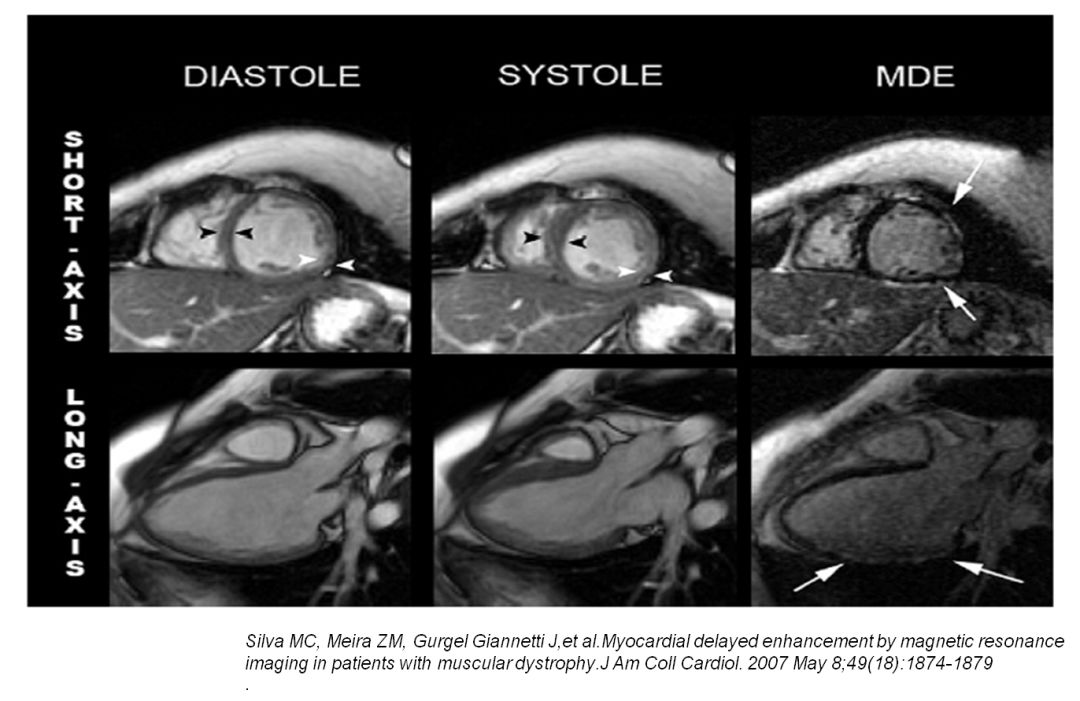
假肥大型肌营养不良病理表现
心肌纤维化和脂肪浸润,尤其以左心室壁明显
假肥大型肌营养不良治疗
无特异性治疗
小剂量强的松可能使疾病发展暂时减慢或增强肌力(0.75mg/kg/天)
呼吸支持,理疗、外科矫形、纠正挛缩,心理治疗
有希望的治疗
成肌细胞移植治疗
基因治疗
病毒载体基因治疗
干细胞治疗
肢带型肌营养不良
(LGMD)
肢带型肌营养不良
发病率:1/14 500 —1/123 000
分类:
LGMD 1( 1A-1F):常染色体显性遗传
LGMD 2( 2A-2I ):常染色体隐性遗传
肢带型肌营养不良发病机制
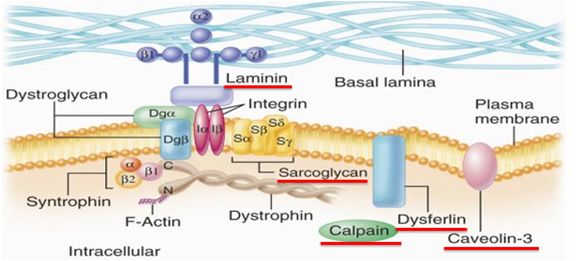
LGMD 主要与一大组肌膜蛋白和近膜蛋白的缺陷有关
LGMD 2D、2E、2C、2F的缺陷蛋白分别为α、β、γ、δ-肌聚糖(sarcoglycan)
LGMD 2A 和2B 的缺陷蛋白分别为calpain 3 和dysferlin
LGMD 1A、1B、1C 的缺陷蛋白分别为myotilin、lamininα 2和caveolin 3
肢带型肌营养不良临床表现
一组临床表现和遗传特点不同的异质性肌病
儿童、青春期或成年时起病
为骨盆带肌和肩胛带肌的肌肉萎缩无力
腓肠肌肥大
部分患者心脏受累
PMD心脏病变治疗
PMD心脏病变
PMD心脏病变处理
通过超声心动图和心电图进行诊断,看是否存在心脏问题
在对心脏进行检查和治疗的同时,进行肺功能的评估和治疗
根据不同患病类型定期检查心脏
在做任何手术前要检查心脏
当心脏射血分数下降时用ACEI/ARB 和β-R 阻断剂进行治疗
必要时进行心脏移植手术或安装心脏起搏器
PMD心脏病变药物治疗
1. ACEI和ARB
推荐所有PMD伴射血分数下降的患者使用ACEI 或ARB(I;B)。
PMD病史≥10年的男孩,在出现射血分数下降前可考虑使用ACEI或 ARB(IIb;B)。
2. β肾上腺素能受体阻断剂
推荐任何PMD伴射血分数下降的患者使用β肾上腺素能受体阻断剂(I;B)。
若无其他指征(如心律失常),目前不推荐在无射血分数下降的情况下使用β肾上腺素能受体阻断剂来延缓或预防扩张型心肌病(III;C)。
3. 盐皮质激素拮抗剂
DMD/BMD伴收缩功能障碍患者,考虑使用醛固酮受体拮抗剂是合理的(IIa;C)。
DMD/BMD伴收缩功能保留患者,特别是有心肌纤维化证据(如CMR的延迟强化)的患者,可以考虑使用醛固酮受体拮抗剂(IIb;C)。
4. 糖皮质激素
DMD患者可考虑使用糖皮质激素来延缓心脏病变的进展(IIb;B)。
其他PMD(包括BMD)患者使用糖皮质激素应视非心脏指征而定(I;C)。
5. 利尿剂
PMD伴心室功能障碍相关体液潴留的患者,应予利尿剂治疗(I;C)。
6. 抗栓药
心室收缩功能正常伴房颤或房扑的PMD儿童,可考虑血栓预防,根据患者血栓风险来确定治疗(IIb;C)。
既往有心律失常病史、心脏受累通常表现为心律失常的PMD患者,不推荐抗凝或抗血小板治疗(III;C)。
7. 抗心律失常药物
除了I,II或IV类抗心律失常药物可能增加外周肌肉无力的警告外,PMD患者使用抗心律失常药与没有PMD的患者相同。应根据每个患者的特殊临床情况进行治疗决策,考虑是否存在传导异常或心肌功能障碍。
PMD心脏病变其他治疗
1. ICD
在部分PMD患者,特别是心律失常可能是主要特征的PMD(DMD,BMD,EDMD,DM1,LGMD1B)患者,考虑ICD是合理的,应进行周密的讨论和决策,个体化考虑(IIa;C)
2. 终末期心衰
对于符合条件的PMD合并终末期心衰患者,可考虑长期机械循环支持(MCS)作为心脏移植的桥接或作为终末治疗(IIb;C)
对于符合条件的PMD、接受了适当治疗仍为终末期心衰的患者,可考虑心脏移植(IIb;C)
对于符合条件的PMD、接受了最佳管理仍为D期心衰的患者,可考虑家中胃肠外应用正性肌力药物作为症状控制的姑息治疗(IIb;C)
病例分析
儿童期发病
临床表现:近端肌肉无力,腓肠肌假性肥大
血清肌酶:升高
肌电图: 肌源性损害
肌肉活检:符合肌营养不良特征
进行性肌营养不良 诊断成立!
BMD? LGMD?
病例分析—家系谱
病例分析—组织活检
肌活检单克隆抗体免疫组化染色:
抗肌萎缩蛋白(dystrophin)-N,-C,-R 表达正常
抗肌聚糖蛋白(sarcoglycan)-α、-β、-γ、-δ表达正常
抗-dysferlin蛋白表达减弱
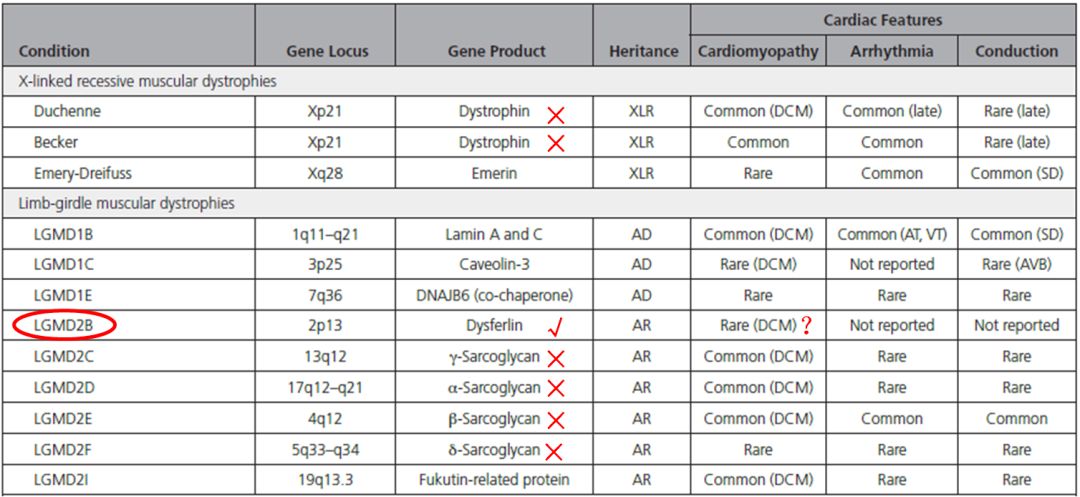
进行性肌营养不良致心脏损害诊断明确
根据肌活检免疫组化结果,考虑为 LGMD2B
进一步需要完善的检查:心肌活检,基因检查
治疗:改善心脏功能
作 者 简 介 :

马慧元
阜外医院心力衰竭中心进修医生
甘肃省人民医院干部病房心内科
医学硕士,心血管内科主治医师。2003年毕业于兰州医学院临床医学系, 同年就职于甘肃省人民医院干部病房心内科。2009年获兰州大学临床医学硕士学位。目前正在阜外医院心力衰竭中心进行专科进修骨干培训。



