
在2017中国国际心力衰竭大会(CIHFC)暨中国医师协会心力衰竭专业委员会年会上,来自北京大学第一医院儿科的杨艳玲教授为我们带来了“儿童代谢性心肌病诊治进展 ”的精彩报告。


◆病种多
— 迄今已发现 900 余种遗传代谢病
◆遗传方式复杂
— 多数常隐,如:
同型半胱氨酸血症
— 少数常显,如:
家族性高胆固醇血症
X连锁遗传,如:Fabry病
线粒体基因遗传,如:MELAS
◆表型复杂
胎儿 - 老年 发病
相同疾病轻重不等
多累及神经系统,多系统损害
◆病因复杂,表型复杂,常规技术难以满足临床需求,必须依赖专业的诊疗技术
◆发生在胚胎 – 成年各个时期,儿科最为多见
◆临床表现复杂,可累及单个脏器,也可能导致多脏器损害,引起神经、肾脏、心血管、血液、消化、皮肤、眼、耳等功能损害,导致残障
◆致死率很高,已经成为我国儿童死亡的主要原因之一
◆随着诊断与治疗学研究的进步,遗传性疾病的病因诊断率显著提高,越来越多的疾病成为可治可防的疾病
◆1.前驱物质异常蓄积,旁路代谢增加,引起代谢紊乱及脑、肝、肾、心脏、骨髓等脏器功能损害。
◆线粒体能量合成功能下降
◆肉碱、生物素等消耗增加
小分子遗传代谢病
— 氨基酸
同型胱氨酸尿症、苯丙酮尿症、枫糖尿症
— 有机酸
甲基丙二酸血症、丙酸血症、异戊酸血症
— 尿素
先天性高氨血症
— 脂肪酸
β氧化异常
— 碳水化合物
半乳糖血症、糖原累积症、果糖不耐受症
— 蛋白
家族性高脂蛋白血症、无白蛋白血症
— 金属
肝豆状核变性、Menkes病
— 嘌呤
雷-尼二氏综合症
— 色素
高铁血红蛋白血症、卟啉病
— 激素
克汀病、先天性肾上腺皮质增生症
◆是一种相对常见的、可治疗的代谢性疾病;
◆病因复杂,与遗传、环境、营养等多种因素有关;
◆导致心、脑、血管、肾脏、眼及骨骼等多脏器损伤;
◆缺乏特异性症状与体征,需要依靠特殊生化检测技术进行筛查与诊断。
同型半胱氨酸血症的病因与类型

◆可以在胎儿 – 成年各个年龄起病;
◆新生儿期多无明显异常,常于婴幼儿时期出现神经精神症状、心血管损害、视力损害等合并症;
◆早期临床诊断困难。
◆神经系统损害:智力运动落后、癫痫、头痛、偏瘫、脑卒中、肌无力,等;
◆骨骼损害:马凡氏综合症样特殊体型,四肢、指(趾)细长,骨质疏松;
◆眼部损害:晶状体脱位、视神经损害;
◆心血管损害:静脉血栓、肺动脉栓塞、心脑血管栓塞、肺动脉高压、心肌病等合并症;
◆其他:大细胞贫血
◆低蛋白饮食,限制蛋氨酸摄入;
◆补充祛除蛋氨酸的特殊奶粉或氨基酸粉;
◆大剂量维生素B6 (100 - 1000 mg/d);
◆甜菜碱(1 – 3 g/d)。
◆保证天然蛋白质供给,保证蛋氨酸摄入;
◆补充叶酸及维生素B12 ;
◆必要时补充蛋氨酸、甜菜碱。
甲基丙二酸尿症合并同型半胱氨酸血症
◆病因: 钴胺素代谢障碍,CblC、CblD、CblF、CblX
◆发病率: 中国人甲基丙二酸尿症中70%以上
◆生化表现:
——甲基丙二酸尿症
——同型半胱氨酸血症 或 同型半胱氨酸尿症
◆药物治疗: 维生素B12、叶酸、左卡尼汀、甜菜碱
◆饮食: 天然饮食
◆疗效: 良好
◆男,2岁后运动发育落后,易疲劳,感冒时咳喘明显,伴口周青紫,手脚皮肤发花。
◆3岁时因“肺炎”入院,发现肺动脉高压,心功能不全,给予药物治疗。
◆4岁后智力倒退,抽搐,伴进行性大细胞性贫血;经血液及尿液生化分析发现“甲基丙二酸尿症合并同型半胱氨酸血症”,开始维生素B12、叶酸、左卡尼汀、甜菜碱治疗;
◆4岁半时重症肺炎、心衰,死亡。
肉碱、脂肪酸代谢障碍与心肌病
◆原发性肉碱缺乏症
◆继发性肉碱缺乏症
◆线粒体脂肪酸代谢障碍
线粒体脂肪酸β氧化障碍
◆肉碱转移酶缺乏症
◆肉碱转位酶缺陷
◆极长链脂肪酸缺乏
◆肉碱棕榈酰转移酶1缺乏症
◆肉碱棕榈酰转移酶2缺乏症
◆极长链脂酰辅酶A脱氢酶缺乏症
◆三头酶缺乏症
◆中链脂酰辅酶A脱氢酶缺乏症
◆短链脂酰辅酶A脱氢酶缺乏症
◆电子传导黄素蛋白缺乏症
肉碱缺乏症的临床表现
◆惊厥,智力运动障碍
◆易疲劳、乏力,活动后加重
◆肥胖或营养不良,便秘
◆抑郁,焦虑
◆脂肪肝,肝肿大,肝损害,酒精不耐受
◆心肌损害,心功能不全,心肌病
◆瑞氏综合征
◆酮症、低血糖
◆高乳酸、高尿酸血症,高氨血症
◆二羧基酸尿症,代谢性酸中毒
◆猝死(心源性)
遗传代谢病 - 细胞器病,小分子代谢病
◆溶酶体贮积症
— 粘多糖病,神经节脑苷脂病,等
◆过氧化物酶体病
— 肾上腺脑白质营养不良,等
◆线粒体病
— Leigh综合征,MELAS
◆合成缺陷
— 蛋白糖基化异常综合征,等
12岁确诊的Fabry病1例
◆男,12岁,家族史(-)
◆4岁起发作性脚趾疼痛,无诱因,不规律,无发热、皮疹、关节肿痛等伴随症状,自然缓解。
◆智力运动正常,营养状况良好,一般体检未见异常。电生理、影像等多种检查未见异常。
◆α-半乳糖苷酶A活性1.0(正常值 24.5 ~ 86.1)nmol/g/min,显著降低。
◆GLA基因突变:IVS6+2T>C
◆治疗:酶替代治疗?细胞移植?
◆结局:肾损害?心血管损害?脑损害?
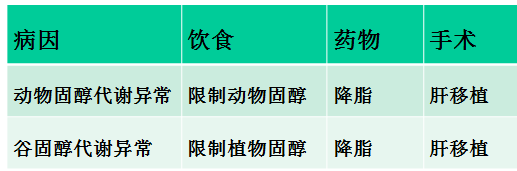
Fabry 病
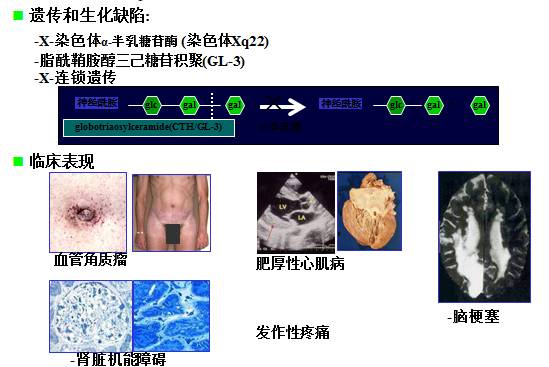
经典Fabry病的自然病程
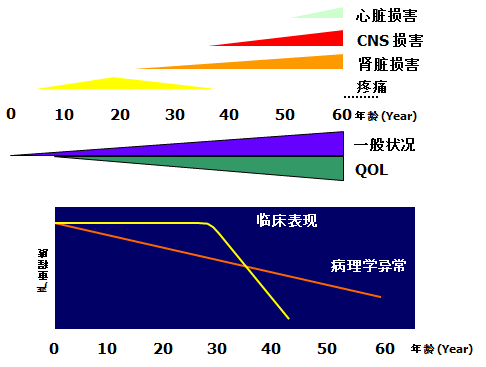
糖原累积症2型(庞贝氏病,Pompe病)
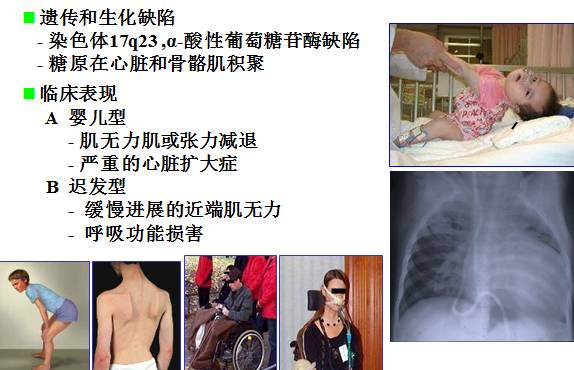
当病人有如下表现时应怀疑 Pompe病
— 婴儿松软伴肥厚性心肌病
— 无法解释的肌肉病变伴呼吸功能障碍
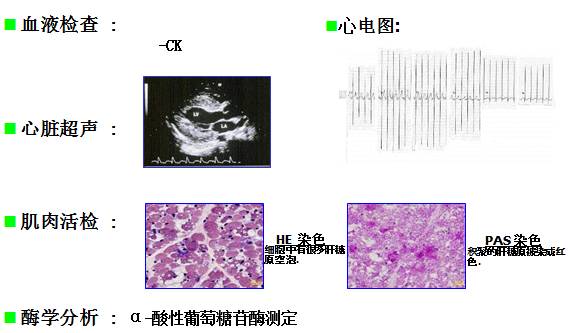
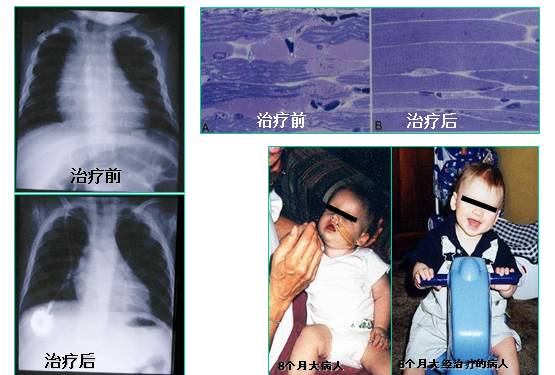
酶替代疗法对婴儿Pompe 病的治疗效果
心肌病, 肌肉病理和运动功能
溶酶体病的治疗方法
◆酶替代治疗
— 糖原累积症2型
— Fabry病
— 戈谢氏病
— ……
◆细胞治疗
— 骨髓移植
— 干细胞移植
◆分子伴侣
◆对症治疗,生活管理
线粒体病Mitochondrial disorders
由于核基因、线粒体基因或两组基因交流缺陷引起线粒体氧化磷酸化障碍,引起脑、心、肌肉、肝、肾等多脏器损害,也称为线粒体细胞病。
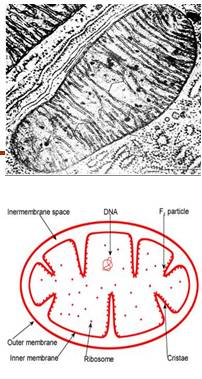
线粒体呼吸链
◆5种酶复合物(I、II、III、IV、V)
复合物II - 由核基因编码
其余复合物- 核基因和线粒体基因共同编码
◆遗传方式复杂:
— 常染色体隐性遗传
— X-连锁遗传
— 母系遗传
— 不明
线粒体病常导致的细胞及组织损害
◆神经损害
— 中枢神经损害
— 末梢神经损害
◆心肌、骨骼肌损害
◆肾损害
◆肝损害
◆营养障碍
线粒体病的诊断方法
◆临床调查:进行性神经、肌肉损害、多脏器损害……
◆化验:血、尿、脑脊液中乳酸、丙酮酸浓度增高;部分患儿伴代谢性酸中毒、血氨增高、低血糖、心肌酶增高、肉碱缺乏……
◆肌肉、神经活检,部分患者可见破碎样红纤维
◆影像学检查:脑MRI(Leigh综合征、MELAS患者可见特异性改变);超声心动图(心肌病)
◆线粒体呼吸链酶活性分析:皮肤成纤维细胞、淋巴细胞、神经细胞或组织
◆基因诊断:线粒体基因或核基因分析
◆解剖诊断:对可疑病例争取尸检
治 疗
◆基本药物
— 维生素 B1 100 - 500 mg/d
— 硫辛酸 200 - 600 mg/d
— 左卡尼汀 500 - 2000 mg/d
— 维生素 E 100 - 300 mg/d
— 维生素 C 100 - 500 mg/d
— 辅酶Q10 20 - 90 mg/d
— 肌酸 1 – 5 g/d
— 其他
◆饮食: 低碳水化合物,中链脂肪酸,蛋白质
应该考虑遗传代谢病的情况
◆间歇性发作的疾病急剧恶化
◆类似中毒但是无毒性物质接触史
◆表现类似严重感染,但是多种感染指标阴性
◆对可疑获得性疾病治疗无反应
◆同胞或近亲曾有类似情况
◆新生儿病情急剧进展,无法解释
◆任何类型的脑病、心肌病,尤其是反复发作性的疾病
◆智力运动发育落后或倒退
◆低血糖
◆瑞氏综合征
◆各种“累积病”(心脏增大,肝大,脾大,骨病…)
遗传代谢病依赖专业的诊断与治疗技术
◆提高警惕,重视症状与体征,发现高危儿
◆一般检查
— 血液:贫血?细胞形态?…
— 尿液:酮体?蛋白尿?…
— 生化:肝肾功能?肌酶增高?低血糖?酸中毒?酮症?…
◆影像学检查:
— 脑CT、MRI:进行性退行性病变?…
— 超声心动图:心肌病
◆肌电图异常?神经传导异常?
◆组织活检:皮肤、肌肉、神经?
◆特殊生化分析
— 尿液有机酸、氨基酸、肉碱、蝶呤
— 血液氨基酸、肉碱谱、脂肪酸
— 血液总同型半胱氨酸、维生素B12、叶酸、生物素…
◆酶学分析:
— 线粒体呼吸链酶复合物活性分析
— 溶酶体相关酶活性分析
— 生物素酶活性分析
◆基因分析
— 疾病相关基因突变分析
◆尸检:化学解剖,组织分析
遗传代谢病筛查技术
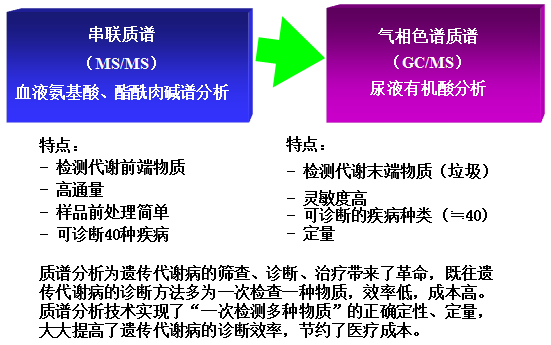
串联质谱血液氨基酸、酯酰肉碱分析
◆1. 典型氨基酸、酯酰肉碱谱,可以确定某些疾病:
— 高苯丙氨酸血症
— 酪氨酸血症
— 瓜氨酸血症1型
— 精氨酸血症
— 异戊酸尿症
— 原发性肉碱缺乏症
…
◆2.某些项目增高或降低,可能提示某种疾病,应在急性期复查,或者采用其他方法鉴别,例如:
— C3 (丙酰肉碱)↑,提示:
— 甲基丙二酸尿症 --- 尿液有机酸分析
— 丙酸尿症 --- 尿液有机酸分析
— 维生素B12、叶酸缺乏症 --- 血液维生素B12、叶酸测定
— 生物素缺乏症 --- 血液生物素、生物素酶测定
— 再甲基化异常
— 精氨酸 ↓,提示:
— 高氨血症2型
— 营养障碍
— 游离肉碱降低,提示:
— 原发性 / 继发性肉碱缺乏 --- 尿液肉碱测定、肾小管功能
◆3.结果正常,不能除外“代谢病”
— 很多疾病只在发作期出现异常,如:
— 高氨血症2型
— 戊二酸尿症2型
— 很多脂肪酸代谢病
— 很多遗传病目前技术尚未涵盖,如:
— 同型半胱氨酸血症2型、3型
— 溶酶体病
— 线粒体病
— 由于仪器或试剂的问题,出现假阳性或假阴性,如:
— 戊二酸尿症1型
◆1. 减少毒性物质来源,减少蓄积
饮食治疗:特殊饮食
◆2. 清除代谢毒物
维生素B12 – 甲基丙二酸尿症
左卡尼汀– 清除有机酸
◆3. 补充生理活性物质
左卡尼汀:促进脂肪酸、有机酸、糖代谢
生物素: 几种羧化酶的辅酶
◆4. 酶替代治疗
溶酶体病
◆5. 细胞移植,器官移植
◆6. 基因治疗
◆预防疲劳
◆注意营养监测,合理干预,保证热量、蛋白质、氨基酸、脂肪酸、维生素的摄入
◆病情控制良好状态下预防接种
作者简介

杨艳玲教授
1984年毕业于北京大学医学部(原北京医科大学医疗系),1996年毕业于日本东京医科齿科大学医学部,获得儿科学博士学位。
◆兼任:
中华医学会儿科分会临床营养学组副组长;
中华预防医学会出生缺陷预防与控制专委会新生儿筛查学组副组长;
中国出生缺陷干预救助基金会专家工作委员会委员、儿童遗传代谢病专家委员会副主任委员;
亚洲遗传代谢病学会委员;
中国医师协会青春期医学专业委员会临床生化学组副组长,临床遗传学组委员;
中国医师协会儿科分会神经修复学组副组长;
中国医师协会检验医师分会线粒体疾病检验医学专家委员会副主任委员;
北京医学会儿科分会内分泌遗传代谢学组副组长;
北京医学会罕见病分会委员。



