
在2016年中国国际心力衰竭大会暨中国医师协会心力衰竭专业委员会第一届学术年会上,来自华中科技大学同济医院心内科郭小梅副教授给我们带来了“肥厚型心肌病诊断与治疗新进展“精彩报告。

肥厚型心肌病(HCM)
经典的HCM发病率约为1/500,临床表型:无症状--胸闷胸痛--猝死。主要遗传方式为常染色体显性遗传,但也有常染色体隐性遗传、伴性遗传、线粒体遗传等多种方式,也不乏新发突变。经典的HCM其发病至少与30余个基因相关。50%-70% HCM为编码肌小节蛋白的基因突变所致。种族不同,突变谱也不同。不同基因受累,同一基因不同突变受累,临床表型和预后不一样。10% HCM患者携带复合致病突变,表型严重程度和突变数量正相关。
HCM患者诊断流程
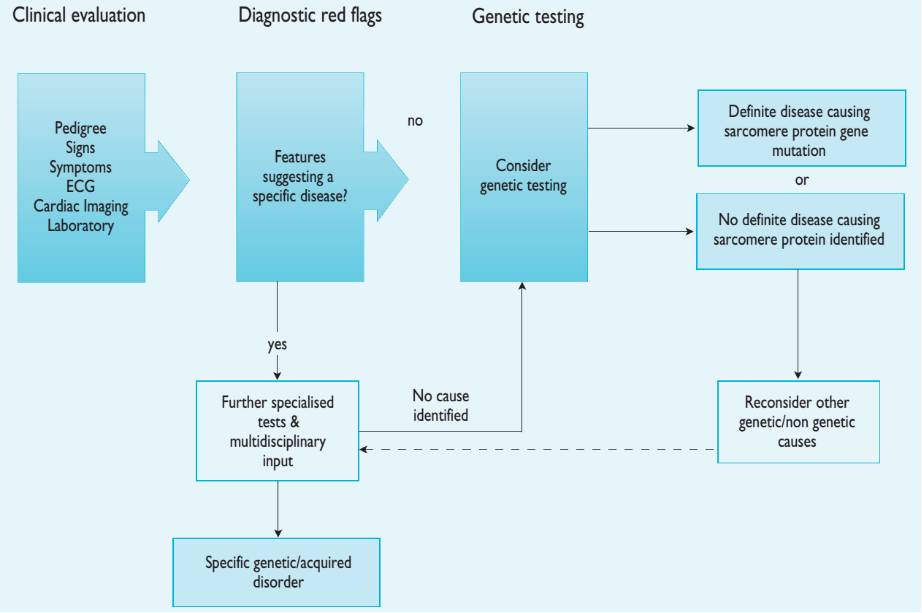
临床诊断
成人HCM 定义:任意成像手段(超声心动图、 心脏核磁共振成像或心脏CT)检测显示,并非完全因心脏负荷异常引起的左室心肌某节段或多个节段室壁厚度≥15 mm。左心室和(或)右心室及室间隔非对称性肥厚为HCM的特征性病理改变。按受累部位不同分为6个亚型:①肥厚局限于室间隔基底段,梗阻常见②心尖部肥厚型③左室前、侧壁肥厚型④左室后壁肥厚型⑤均匀肥厚型⑥右室肥厚型。
超声心动图
对所有初诊的HCM患者都推荐行经胸超声心动图检查,推荐从基底到心尖用二维短轴切面测量每个左室节段的最大舒张期室壁厚度(≥15 mm) 。左室流出道梗阻(LVOTO)定义为静息或生理激发状态下多普勒超声测量的瞬时最大左室流出道压力阶差≥30mmHg。通常≥50mmHg才有临床意义。在室间隔消融术前或怀疑存在其他瓣膜病变时,经食管超声心动图可用于经胸视野较差的患者或者作为心脏磁共振的补充,以确定LVOTO的机制。
心脏磁共振(CMR)和延迟轧显像(LGE)

侵入性血流动力学研究建议

心内膜活检 若其他临床检查提示存在心肌浸润或炎症,且没有其他验证手段,可考虑进行心内膜心肌活检。(IIb C)
HCM鉴别诊断
代谢或浸润蓄积病:6种 (预后均极差)
1)线粒体病
2)Fabry病(α-半乳糖苷酶缺乏)
3)糖原蓄积病: PRKAG2(一磷酸腺苷激活的蛋白激酶γ调节亚单位-2)
4)LAMP2(溶酶体相关膜蛋白基因-2,Danon病)
5)Noonan综合征: RAS病(RASopathy)
6)Pompe病( 糖原蓄积病II,缺乏α1,4-酸性麦芽糖苷酶)
其他鉴别诊断:
1)运动员心脏 (预后好)
2)高血压心脏肥厚 (预后好)
遗传学检测和家族筛选
约2/3的HCM患者表现出家族性遗传特点,目前报道HCM相关基因突变己超过900种,由MYH7、MYBPC3和TNT等3个基因突变引起的HCM约占3/4。
HCM先证者及亲属遗传检测流程图
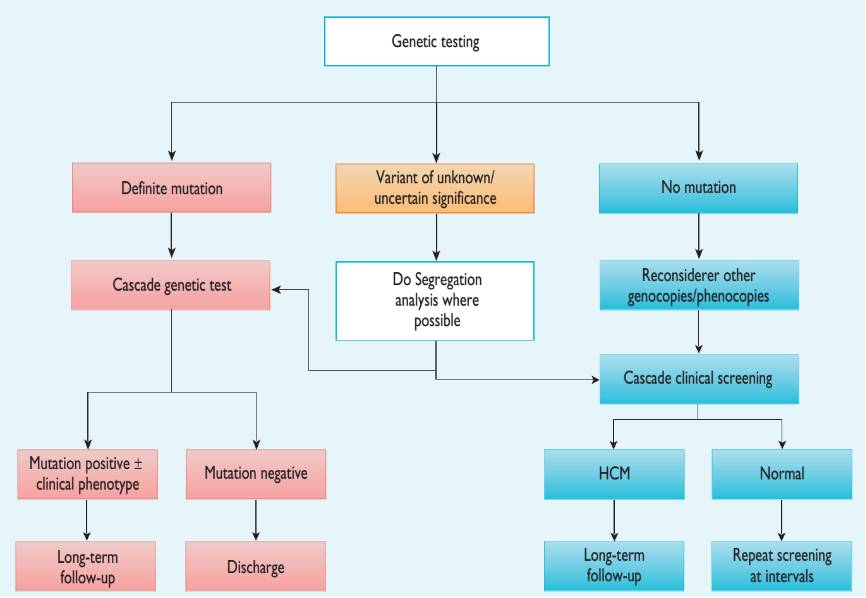
对于先证者遗传检测的建议

成年亲属遗传和临床检查推荐

对儿童进行遗传检测和临床检查的建议


最常见的两个致病基因
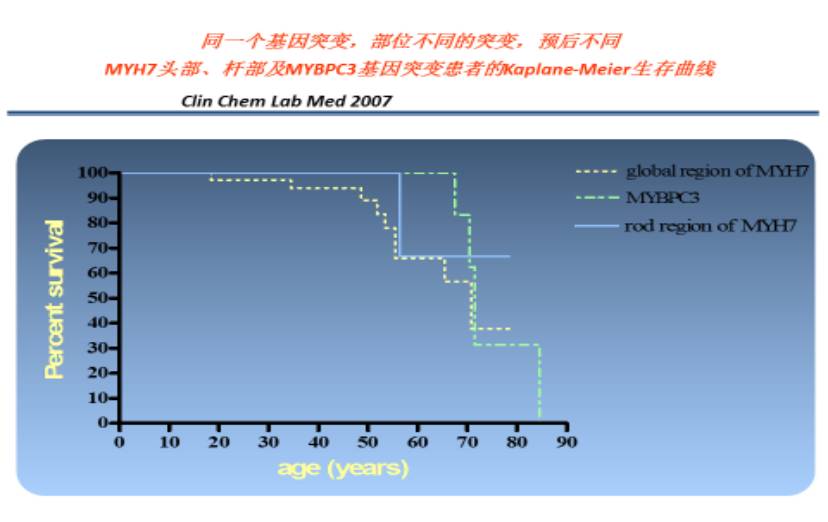
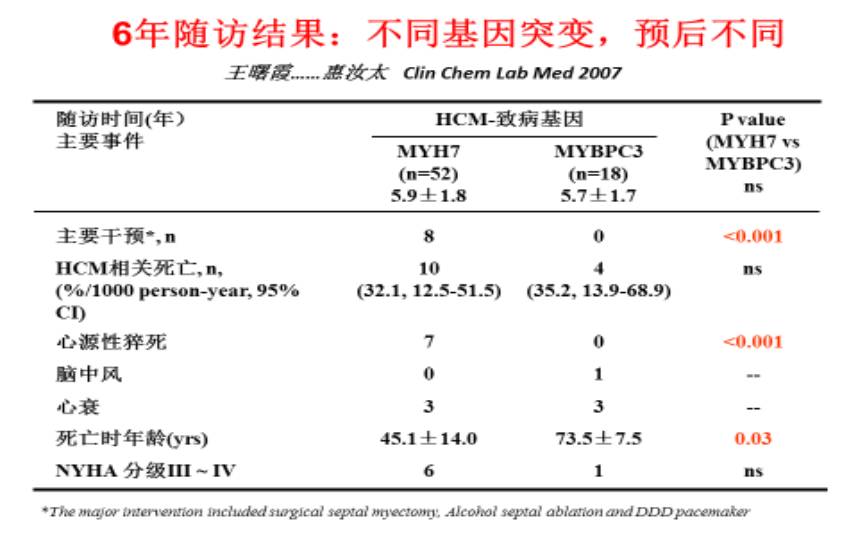
心肌病基因诊断的意义
帮助临床明确诊断:
1. 继发性心肌损害 or 原发性遗传性心肌病?
2. 单纯心肌病 or 包含心肌损害的多器官综合征?
3. 帮助精确定位疾病阶段:是扩张型心肌病 or 肥厚型心肌病扩张期?
帮助临床判断预后:
1. 良性基因(如:MYBPC3)&良性突变(如:错义突变)受累预后较好(药物治疗基本可以满意)
2. 恶性基因 (如:PRKAG2等代谢性疾病致病基因)&恶性突变 (如:移码突变、无义突变)受累预后较差(高猝死风险,需积极考虑装ICD或心脏移植)
需积极考虑特殊治疗的HCM类型
无论哪个基因,当出现严重的LOF突变(框移突变、无义突变、起始密码子突变)或纯合子突变时,患者临床上病情进展快,提示预后差,需积极考虑心脏移植
MYH7、TNNT2、TNNI3三个基因导致的HCM常合并快速性恶性心律失常,易发生猝死,需积极考虑ICD。合并糖原累积病
6种代谢病导致的心肌肥厚,通常预后很差,尤其是:
1.PRKAG2基因:患者临床表现为心肌肥厚合并预激综合征,心动过缓,
易心跳骤停猝死,应积极考虑起搏器
2.LAMP2基因:患者临床早期肥厚,之后扩张,快速进展为心衰,需积
极考虑心脏移植
3.淀粉样变性 & 糖原累积病等代谢疾病导致的心肌肥厚仅是全身性疾病的一部分,
病情重,进展快,应优先考虑心脏移植
WPW综合征,ARVC有高猝死风险,需要安装ICD
心肌病基因诊断的意义
帮助优生优育:
1. 常染色体显性遗传模式的心肌病(MYH7基因突变)患者兄弟姐妹与后代患病概率50%
2. 常染色体隐性遗传模式的心肌病(TAZ基因突变)患者的兄弟姐妹与后代至少是携带者
3. 产前诊断可以在怀孕早期判断胎儿是否携带致病突变
4. 试管婴儿技术结合胚胎单细胞测序可以实现后代不携带致病突变
相对较为缓和的HCM类型
突变类型为杂合错义突变,突变发生在基因的非重要功能区域,或多个重复拷贝的可替换区域,通常对基因功能影响不是太大,患者临床症状轻或者无症状。当致病基因为MYBPC3时,突变类型为非LOF杂合突变,通常患者发病年龄比较大,仅表现为心肌肥厚,病情进展缓慢,猝死风险比较低。值得一提的是,如果是MYBPC3基因发生的LOF突变,患者通常会在心肌肥厚之后开始进入扩张期,随后发生心衰,但预后仍较其他基因发生LOF突变的患者要好。
心肌病检测套餐--80个致病基因
病因
肥厚型心肌病
限制性心肌病
扩张型心肌病
致心律失常右室心肌病
心肌致密化不全
所有鉴别诊断的遗传病
“症状导向型”设计,针对所有心肌病症状的疾病,检测结果结合临床表现可以指导临床精准分型,精准治疗。
所有心肌病患者需提供:
普通心电图
长程心电图
心脏彩超/心脏CT/心脏MRI报告(最好能区分梗阻与否)
血生化指标(cTnI、BNP、CK、K、Ca、Mg、乳酸)
猝死家族史问诊,高血压病史,糖尿病史问诊
详细的晕厥史问诊情况(晕厥次数、晕厥时间、持续时间、有否意识完全丧失、有否大小便失禁、有否手足抽搐、有否口吐白沫
病例1.
患者,男,29岁,胸闷心悸3月入院,既往有2次晕厥史,不伴四肢抽搐,口吐白沫,大小便失禁
心脏彩超示:左室后壁增厚(14mm),心房心室不扩大,心功能正常
普通心电图示:窦性心动过缓55bpm,左心室肥大?
长程心电图提示:预激综合征?
初步临床诊断:1.肥厚型心肌病;2.心律失常
基因诊断结果:
并非是HCM和心律失常两种疾病,而是一个综合征!
不能仅单纯按普通HCM药物治疗,患者高猝死风险,需考虑安装ICD!
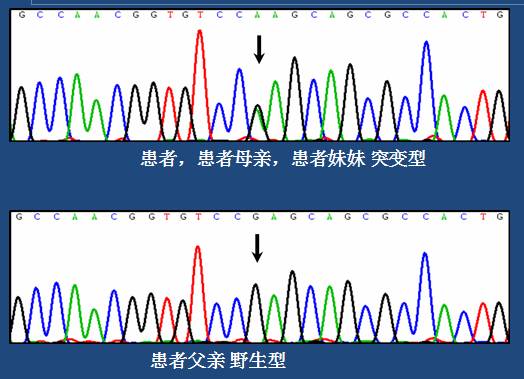
PRKAG2基因第7外显子存在c.905G>A杂合突变,导致该基因编码蛋白发生p.Arg302Gln错义突变,该突变为已报道过的Wolff-Parkinson-White综合征(糖原累积病)致病突变rs121908987(PMID:11407343)。患者其他亲属(母亲、妹妹)该位点亦为突变型,患者父亲该位点则为野生型。
病例2:被隐藏了的“致命性ST段抬高”
11岁男性患者,因“体检发现ECG胸导联ST段抬高”前来就诊,无明显症状,无晕厥史,无猝死家族史
心脏彩超提示:心肌非对性肥厚,最厚处14mm
基因诊断提示:同时患有肥厚型心肌病和Brugada综合征(考虑新发突变)
补行激发试验:阳性结果
肥厚型心肌病患者胸前导联ST段抬高----不一定致命
Brugada综合征患者有时会有胸前导联ST段抬高----致命!
同时患有肥厚型心肌病的Brugada综合征的患者极容易被忽略!

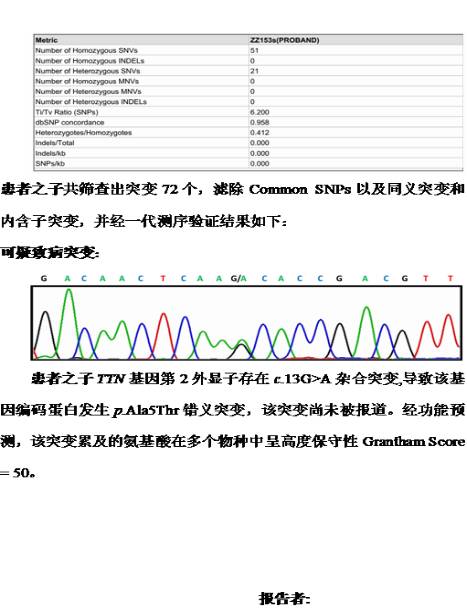
病例3.致病突变有时未必“一脉相承”

HCM患者合并心力衰竭诊治流程---2014ESC
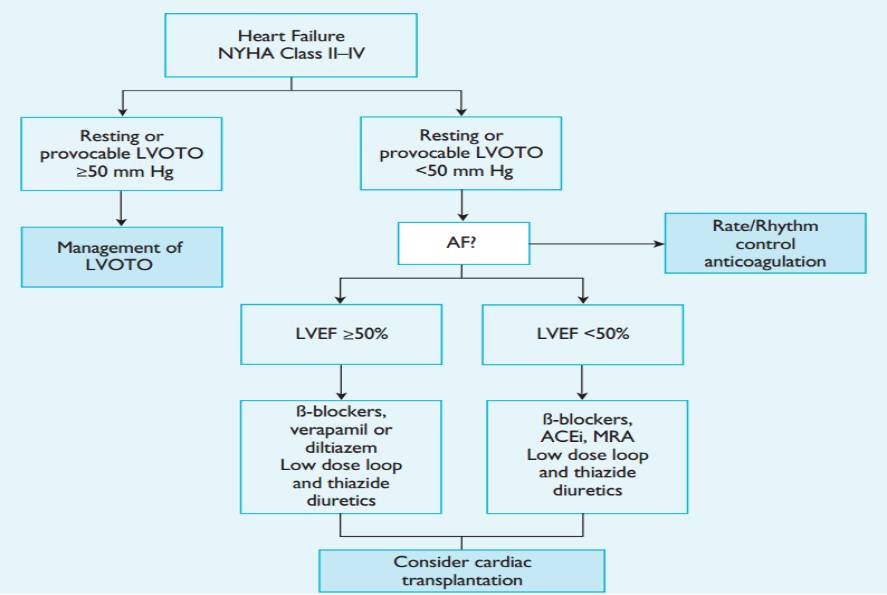
左室流出道梗阻(LVOTO)治疗

LVOTO药物治疗建议

LVOTO室间隔减容治疗建议


LVOTO起搏治疗建议

EF≥ 50%的心衰患者治疗建议

EF<50%患者治疗建议


心脏移植建议

左室辅助装置建议




