

➤ 扩张型心肌病
• DCM是一种异质性心肌病,以心室扩大和心肌收缩功能降低为特征。表现为:心脏逐渐扩大、心室收缩功能降低、心衰、室性和室上性心律失常、传导系统异常、血栓栓塞和猝死。
具有心室扩大和心肌收缩功能降低的客观证据:
1. LVEDd >5.0cm(女性)和LVEDd >5.5cm(男性)(或大于年龄和体表面积预测值的117%,即预测值的2倍SD+5%)
2. LVEF<45%(Simpsons法),LVFS<25%
3. 发病时除外高血压、心脏瓣膜病、先天性心脏病或缺血性心脏病
➤ 扩张型心肌病的病因分类 —— 病因分类
• 继发性
➤ 家族性扩张型心肌病(FDCM)
• 上世纪八十年代以前,认为只有1%~2%的DCM病例有家族倾向,仅有散在的家族性病例报道。
• 随着对DCM认识和诊断手段的进步,发现越来越多的DCM患者有遗传学基础,呈现家族聚集趋势,这部分患者也称为“家族性扩张型心肌病”(familial dilated cardiomyopathy,FDCM)。
• FDCM是由于特定的基因突变导致其编码的心肌细胞蛋白分子异常,并最终导致心脏结构和功能损害。
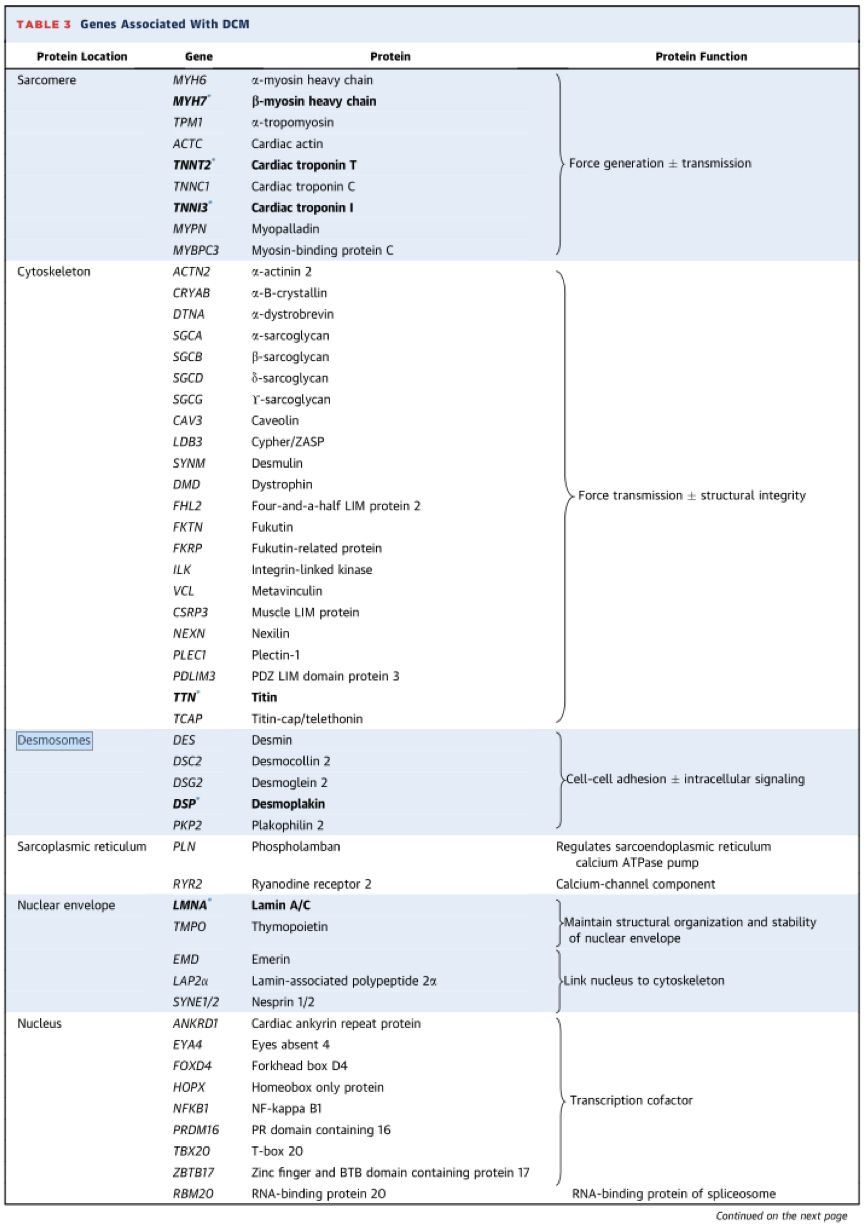
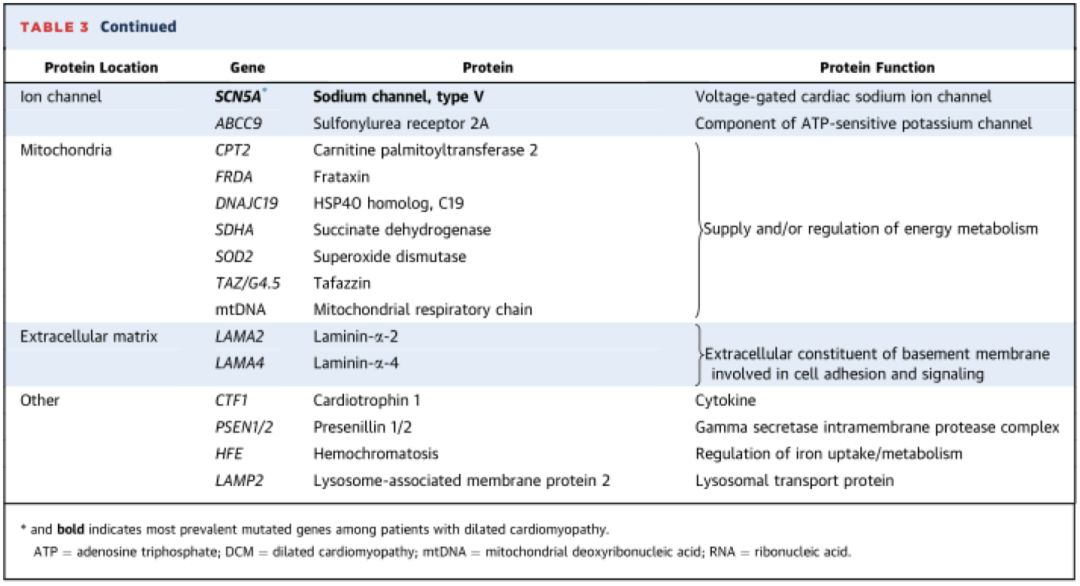
• FDCM致病基因大多编码细胞骨架和/或收缩成份的蛋白,包括肌营养不良蛋白、心肌肌动蛋白、结蛋白、核纤层蛋白及离子通道蛋白等。
• FDC相关的基因突变主要影响心肌细胞的能量产生、能量传输、机械收缩和信号转导。
• 同一家族的一级亲属中有2例(包括先证者)及2例以上DCM患者
• 或DCM患者的一级亲属有尸检证实为DCM。
• 或不明原因的50岁以下猝死者。
• 研究发现FCDM在DCM中的发病率占25-50%,真实的比率应该会更高。
➤ 家族性DCM的遗传特点
1. 遗传异质性:不同基因的不同突变可导致同样的FDCM表型,同一家族相同基因的同一突变位点可产生不同表型。
2. 基因突变外显不全:外显率会随着年龄的增大而增高,常染色体显性遗传者在<20岁时外显率为10%,20-30岁者为20% ,30-40岁者为50%,>40岁时外显率达到90%。
3. 遗传方式多样性:包括常染色体显性遗传、常染色体隐性遗传、X-连锁染色体遗传及线粒体遗传。FCDM以常染色体显性遗传最为常见。
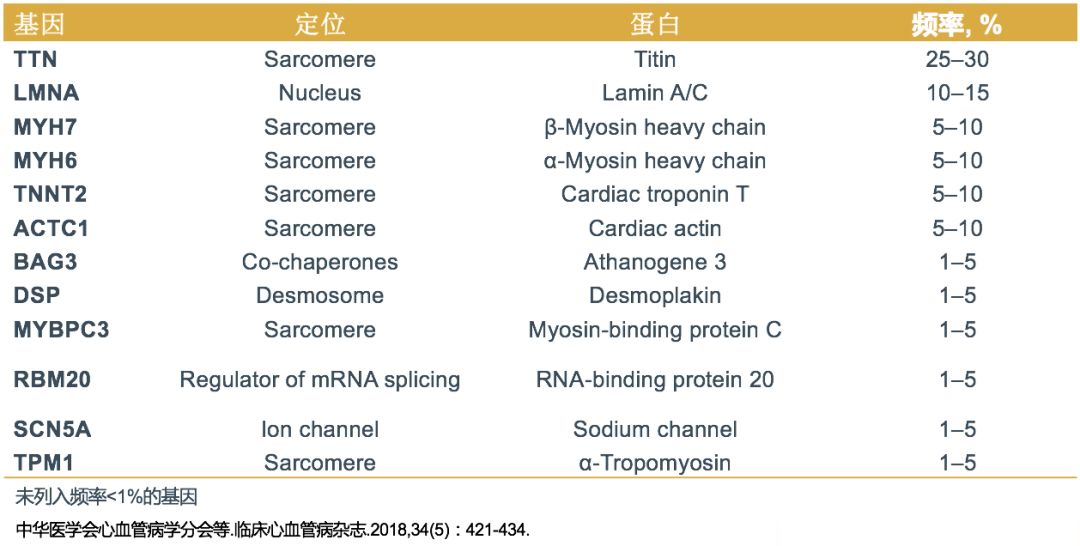
➤ 影响肌小节的基因突变
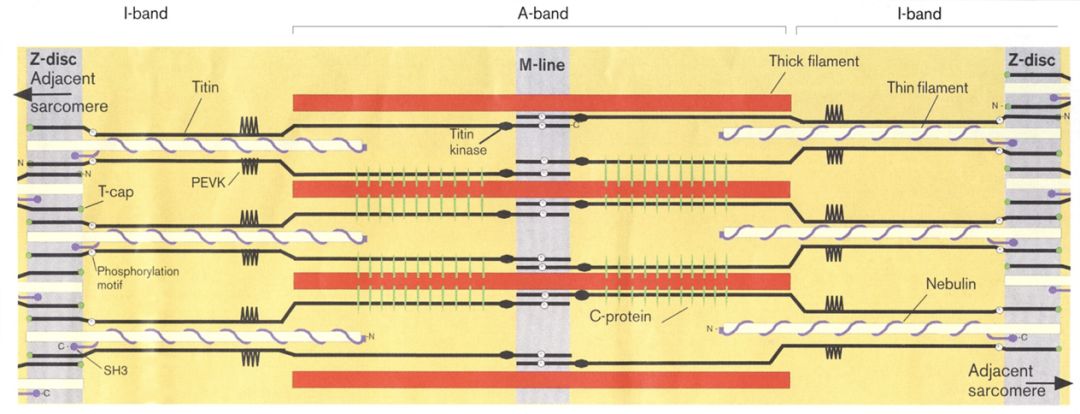
• TTN是DCM最常见的致病基因,约占DCM致病基因的25%。
• 患者通常于40岁之前出现典型的DCM表现。
• TTN编码肌联蛋白,是粗、细肌丝相互作用的支撑点,在肌小节组装和原力产生中发挥重要作用。
• TTN基因的致病突变主要是截断突变,其次为错义突变。
• TTN基因突变蛋白不能发挥生物学作用,从而表现出心脏收缩功能减低。
• TTN突变的患者心律失常风险和心功能失代偿程度更为严重。
• ACTC1基因编码α-肌动蛋白。
• α-肌动蛋白存在于肌肉组织中,是肌肉收缩单位的主要组成部分。
• 目前发现7种ACTC1病理性突变与DCM相关,其中6种为错义突变,1种为剪接突变。
• 核酸蛋白可以结合于心肌细胞核区域,调控核-细胞骨架蛋白相互作用,对于稳定染色质结构、调控细胞转录发挥重要作用。
• 目前发现了3种核酸蛋白与家族性DCM相关,即LMNA、TMPO和EMD,其中EMD是伴X染色体遗传。
• TMPO基因编码促胸腺生成素蛋白,该蛋白参与核纤层蛋白组装,促进核被膜结构稳定。
• LMNA基因编码核纤层A/C蛋白。核纤层蛋白是位于核膜内层的一种纤维结构,与内膜整合蛋白相互作用稳定核孔复合物。
• LMNA基因是DCM第2种主要致病基因,占DCM基因突变类型的10-15%,通过常染色体显性遗传。
• 目前发现了100+种LMNA基因突变类型。
• LMNA突变致病的DCM外显率极高,患者常于中年之前发病,并且在60岁之前临床症状完全出现。
• 该基因突变的临床症状各不相同,常累及心脏传导系统(窦缓、AVB、房性快速性心律失常),骨骼运动受累,最终发展为心力衰竭。
• 约70%的患者于诊断后5年内出现恶性心脏事件,因此该类基因突变的患者建议植入ICD或行心脏移植。
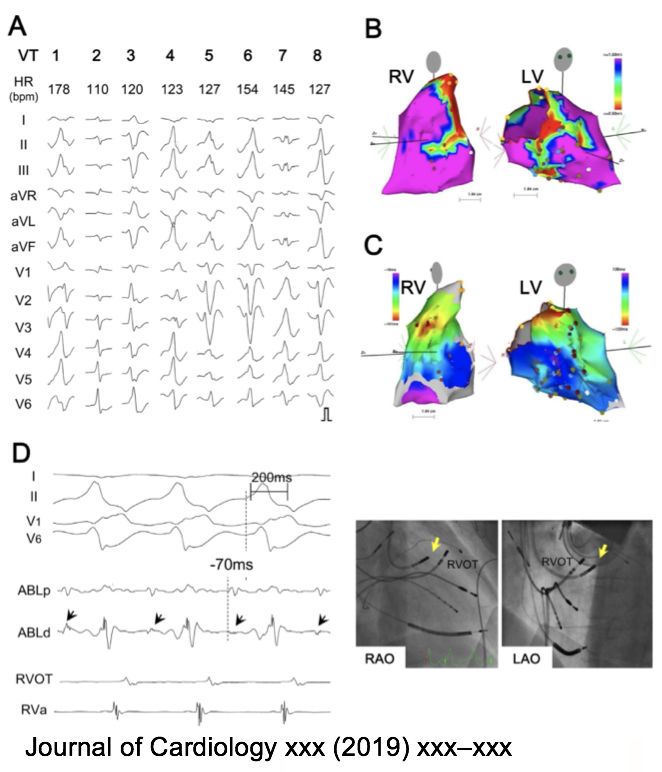
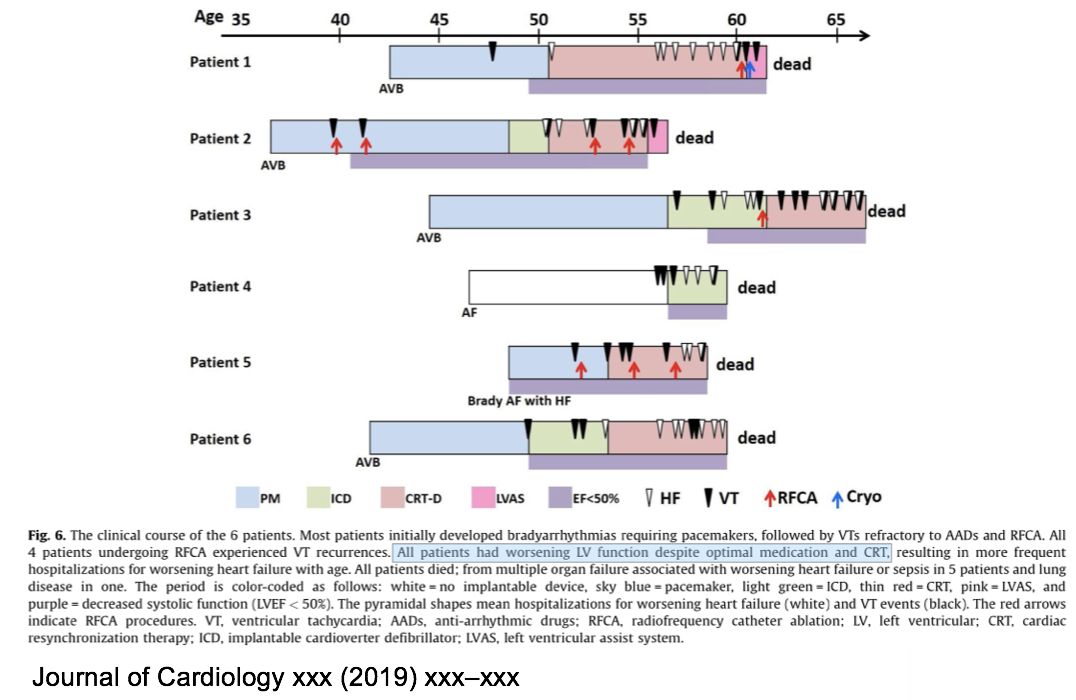
• 6例lamin A/C gene (LMNA) mutation患者,来自3个家系。
• 第一次发作室速的平均年龄为50岁,室速的形态基本一致。
• 所有患者室速射频消融之后平均4个月复发,可能由于室间隔深处病灶。
• 病情进展快,预后差。所有患者因为频发室速、进行性心衰死亡,平均死亡年龄59.5 岁。
• 结论:应该考虑对LMNA基因突变的DCM患者在50岁之前进行心脏移植。
➤ 影响离子通道的基因突变
• SCN5A基因编码钠离子通道的α亚单位。钠通道参与动作电位传导,SCN5A基因突变会扰乱动作电位传导,影响心肌细胞收缩,导致DCM。
• KCNQ1基因与钾离子通道相关,ABCC9基因与ATP结合蛋白相关。
• 肌浆网是细胞储存和释放钙离子的场所。
• PLN基因编码肌浆网的受磷蛋白,PLN基因突变后,心肌细胞舒缩功能障碍。
➤ 转录因子基因突变
➤ BAG3 基因突变
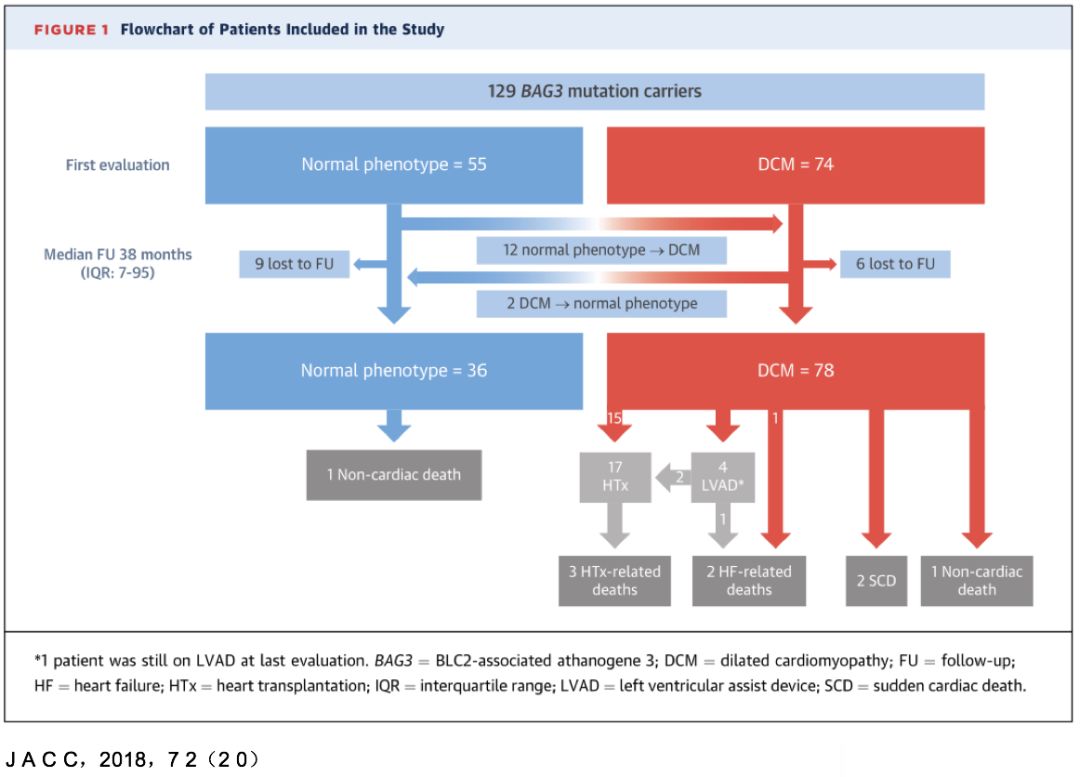
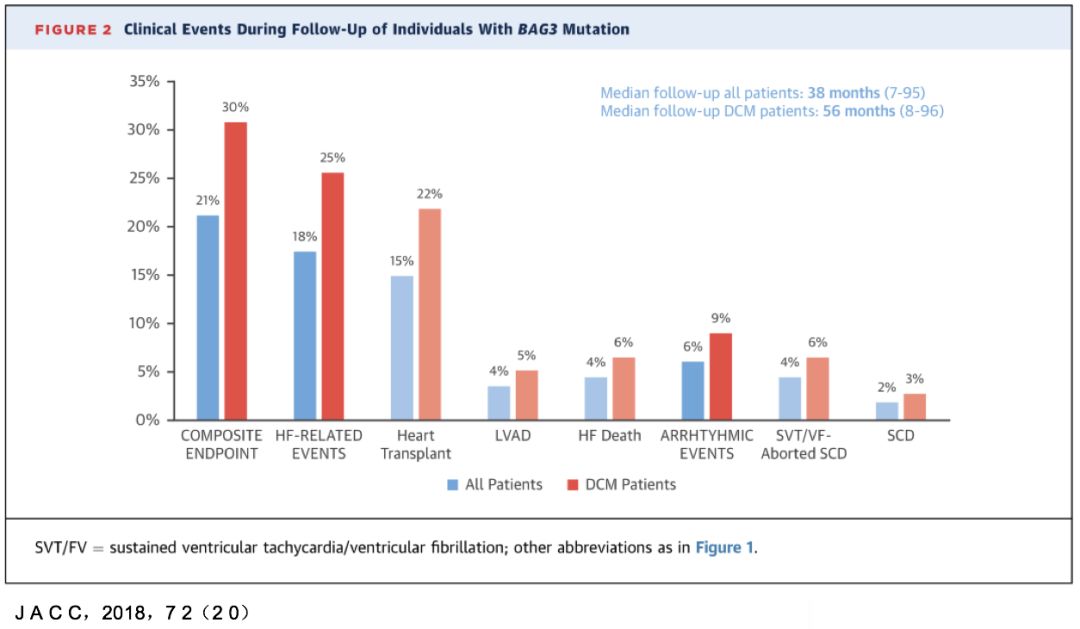
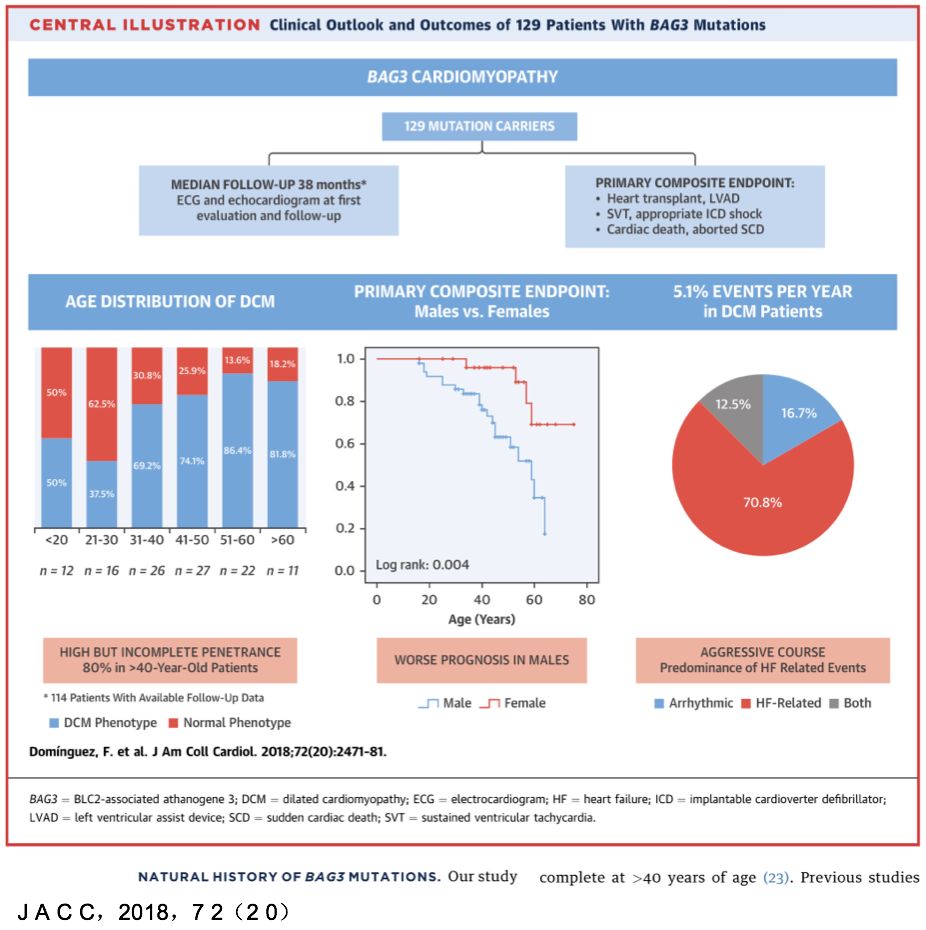
• 首次访视:57.4%为DCM
• 平均随访3.5年后,68.4% 为DCM,其中26.1% 最初心脏表现正常。
• 最后一次访视:Bag3基因突变的外显率为80%,男性发病更早。
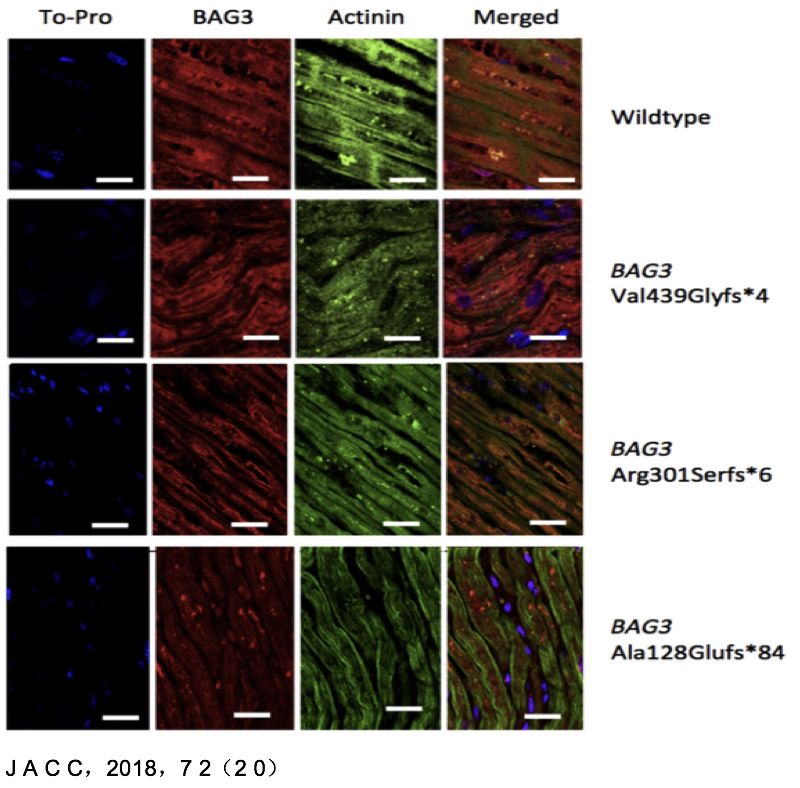
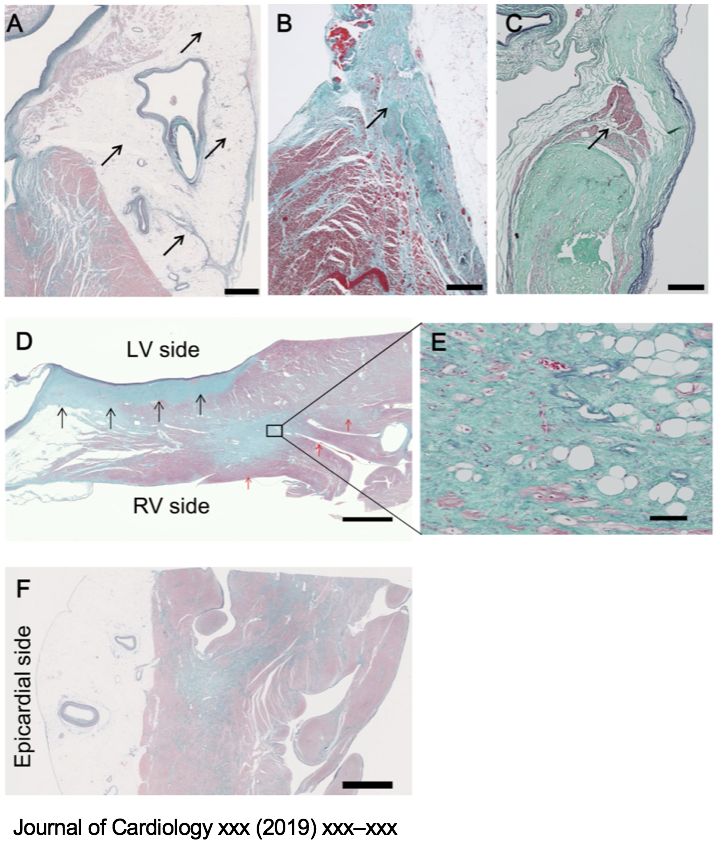
Bag3突变患者的心肌组织显示肌原纤维紊乱。
• 建议对DCM患者亲属进行初级筛查,采集包括患者在内的至少3代亲属的家族史,同时对一级亲属行临床评估。
• 通过心电图和超声心动图进行初级筛查,必要时心脏核磁共振成像检查。其次是采集心律失常及神经肌肉系统疾病病史。
• 研究发现,在临床初筛中10%伴有中度心脏超声异常的患者会在5年内发展为心肌病。
• 2016年AHA关于DCM的科学声明,无论患者是否患有家族性特发性DCM,均应行基因检测(A类证据)。
• 2016年ESC建议,所有家族性或非家族性DCM若出现临床线索,如房室传导阻滞或肌酸激酶升高,均应行基因检测。但ESC强调基因检测应当以临床诊断为前提,并严格限定检测的基因为已知DCM致病基因。
• 一套标准的DCM基因检测板包括40~50个基因,目前新一代测序板检测家族性DCM的阳性率可达40% 。
➤ 遗传咨询
遗传咨询是为了给患者及其家属提供有效信息,包括基因风险、疾病结局、遗传可能性、疾病管理以及计划生育。以专业遗传咨询人员为主导,咨询内容主要包括对可能基因检测结果的讨论,遗传风险的评估以及进行家族性测试。
➤ FDCM家族成员可以分为受累、未受累和不确定三类
• 受累个体符合DCM诊断标准。
— 节段性室壁运动异常(超过一个节段),无缺血性心肌病和室内传导异常。
➤ DCM基因检测尚未全面普及的原因
• DCM是一种高度遗传异质性的心肌疾病,受基因外显率和环境因素等的影响。临床上基因突变携带者的临床表现非常多样、非常复杂。
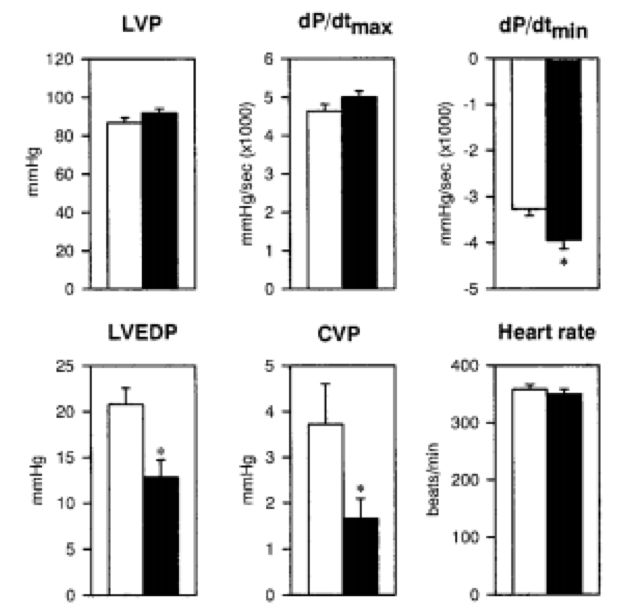
30周后两组比较,黑色代表+delta SG基因治疗
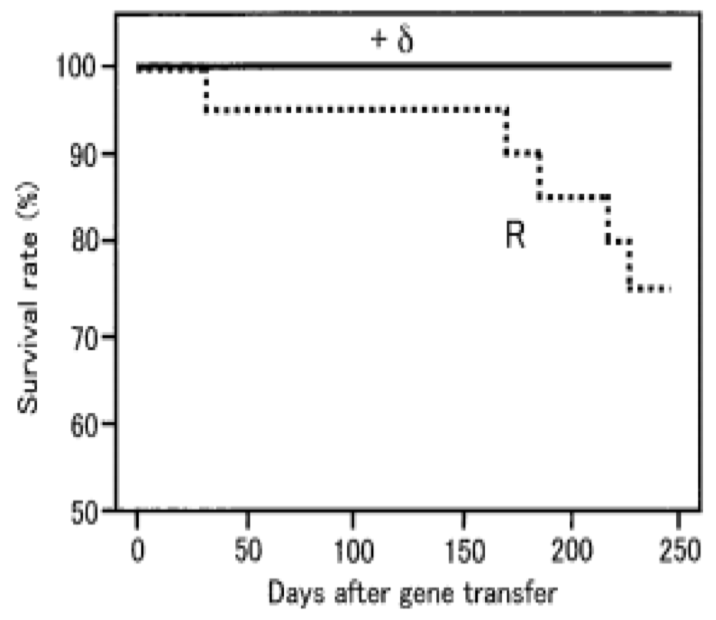
Kawada T,Proc Natl Acad, 2002
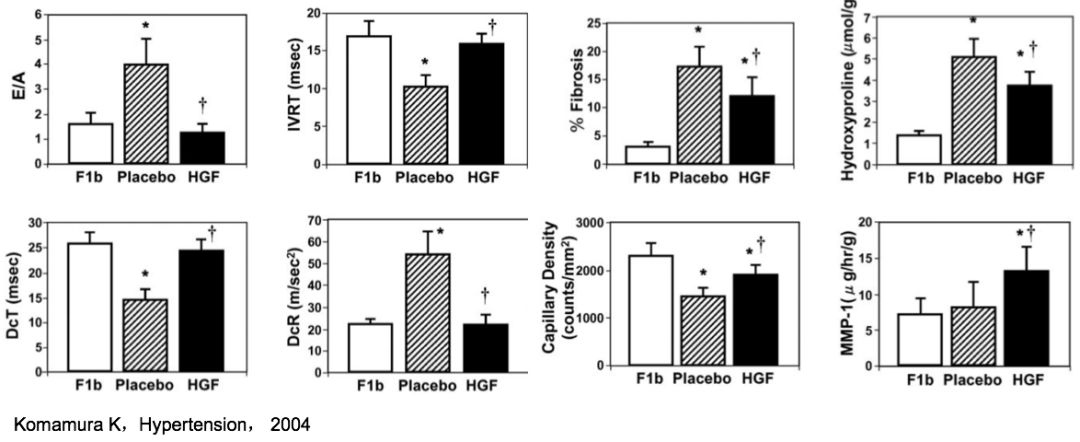
F1B:未经任何治疗的仓鼠;Placebo:安慰剂组;HGF:肝细胞生长因子*P<0.05 vsF1b. †P<0.05 vs placebo.
• 除家族史外,尚无临床或组织病理学检查以判断DCM患者是否为家族遗传性;
• 由于受到表型复杂、外显率随年龄增加的影响,由于家族史不完善,易导致一些家族性DCM被误认为散发病例,DCM通过早期临床表现进行筛查FDCM比较困难;
• DCM在遗传上具有异质性,同一家族的同一基因突变可导致不同的表型,而不同基因突变可导致相同的临床表型,这与不同的环境因素和修饰基因等有关。
王江教授
陆军军医大学附属新桥医院




